Takeda Jun-ichi, Suzuki Yutaka, Nakao Mitsuteru, Kuroda Tsuyoshi, Sugano Sumio, Gojobori Takashi, Imanishi Tadashi
Integrated Database Group, Japan Biological Information Research Center, Japan Biological Informatics Consortium, AIST Bio-IT Research, Building Aomi 2-42, Koto-ku, Tokyo 135-0064, Japan.
Nucleic Acids Res. 2007 Jan;35(Database issue):D104-9. doi: 10.1093/nar/gkl854. Epub 2006 Nov 27.
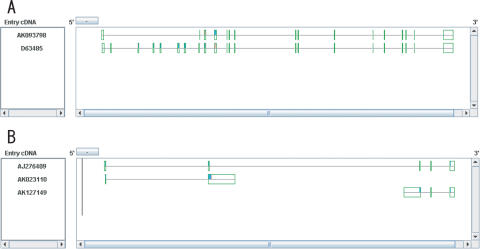
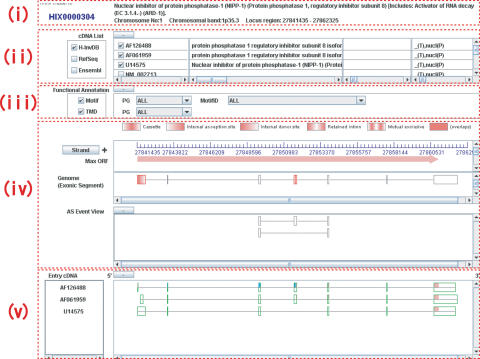
The Human-transcriptome DataBase for Alternative Splicing (H-DBAS) is a specialized database of alternatively spliced human transcripts. In this database, each of the alternative splicing (AS) variants corresponds to a completely sequenced and carefully annotated human full-length cDNA, one of those collected for the H-Invitational human-transcriptome annotation meeting. H-DBAS contains 38,664 representative alternative splicing variants (RASVs) in 11,744 loci, in total. The data is retrievable by various features of AS, which were annotated according to manual annotations, such as by patterns of ASs, consequently invoked alternations in the encoded amino acids and affected protein motifs, GO terms, predicted subcellular localization signals and transmembrane domains. The database also records recently identified very complex patterns of AS, in which two distinct genes seemed to be bridged, nested or degenerated (multiple CDS): in all three cases, completely unrelated proteins are encoded by a single locus. By using AS Viewer, each AS event can be analyzed in the context of full-length cDNAs, enabling the user's empirical understanding of the relation between AS event and the consequent alternations in the encoded amino acid sequences together with various kinds of affected protein motifs. H-DBAS is accessible at http://jbirc.jbic.or.jp/h-dbas/.
人类可变剪接转录组数据库(H-DBAS)是一个专门收录人类可变剪接转录本的数据库。在该数据库中,每个可变剪接(AS)变体都对应一个经过完全测序且精心注释的人类全长cDNA,这些cDNA是为H-Invitation人类转录组注释会议收集的。H-DBAS总共包含11744个基因座中的38664个代表性可变剪接变体(RASV)。数据可通过可变剪接的各种特征进行检索,这些特征是根据人工注释进行标注的,例如可变剪接模式、编码氨基酸的相应变化以及受影响的蛋白质基序、基因本体(GO)术语、预测的亚细胞定位信号和跨膜结构域等。该数据库还记录了最近发现的非常复杂的可变剪接模式,其中两个不同的基因似乎存在桥接、嵌套或退化(多个编码序列)的情况:在所有这三种情况下,一个基因座编码完全不相关的蛋白质。通过使用可变剪接查看器,可以在全长cDNA的背景下分析每个可变剪接事件,从而让用户凭经验了解可变剪接事件与编码氨基酸序列的相应变化以及各种受影响的蛋白质基序之间的关系。可通过http://jbirc.jbic.or.jp/h-dbas/访问H-DBAS。