Sonego Paolo, Pacurar Mircea, Dhir Somdutta, Kertész-Farkas Attila, Kocsor András, Gáspári Zoltán, Leunissen Jack A M, Pongor Sándor
Protein Structure and Bioinformatics Group, International Centre for Genetic Engineering and Biotechnology, Padriciano 99, 34012 Trieste, Italy.
Nucleic Acids Res. 2007 Jan;35(Database issue):D232-6. doi: 10.1093/nar/gkl812. Epub 2006 Nov 16.
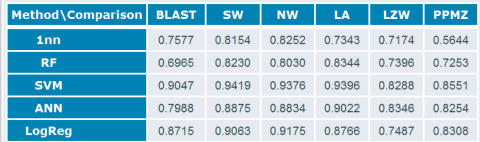
Protein classification by machine learning algorithms is now widely used in structural and functional annotation of proteins. The Protein Classification Benchmark collection (http://hydra.icgeb.trieste.it/benchmark) was created in order to provide standard datasets on which the performance of machine learning methods can be compared. It is primarily meant for method developers and users interested in comparing methods under standardized conditions. The collection contains datasets of sequences and structures, and each set is subdivided into positive/negative, training/test sets in several ways. There is a total of 6405 classification tasks, 3297 on protein sequences, 3095 on protein structures and 10 on protein coding regions in DNA. Typical tasks include the classification of structural domains in the SCOP and CATH databases based on their sequences or structures, as well as various functional and taxonomic classification problems. In the case of hierarchical classification schemes, the classification tasks can be defined at various levels of the hierarchy (such as classes, folds, superfamilies, etc.). For each dataset there are distance matrices available that contain all vs. all comparison of the data, based on various sequence or structure comparison methods, as well as a set of classification performance measures computed with various classifier algorithms.
通过机器学习算法进行蛋白质分类目前已广泛应用于蛋白质的结构和功能注释。蛋白质分类基准数据集(http://hydra.icgeb.trieste.it/benchmark)的创建是为了提供标准数据集,以便能够比较机器学习方法的性能。它主要面向那些有兴趣在标准化条件下比较方法的方法开发者和用户。该数据集包含序列和结构的数据集,并且每个数据集都以多种方式细分为正/负、训练/测试集。总共有6405个分类任务,其中3297个是关于蛋白质序列的,3095个是关于蛋白质结构的,10个是关于DNA中蛋白质编码区域的。典型任务包括基于序列或结构对SCOP和CATH数据库中的结构域进行分类,以及各种功能和分类学分类问题。对于层次分类方案,分类任务可以在层次结构的不同级别(如类、折叠、超家族等)上定义。对于每个数据集,都有距离矩阵可供使用,这些矩阵包含基于各种序列或结构比较方法的所有数据之间的全对全比较,以及使用各种分类器算法计算的一组分类性能度量。