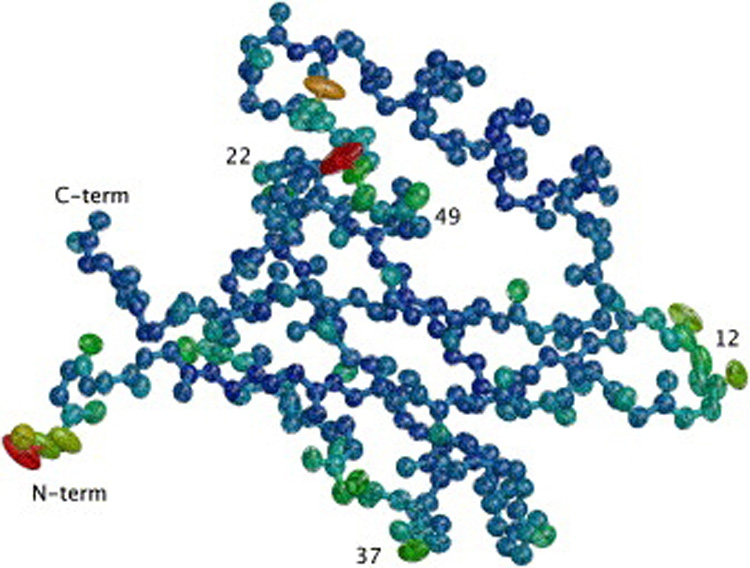
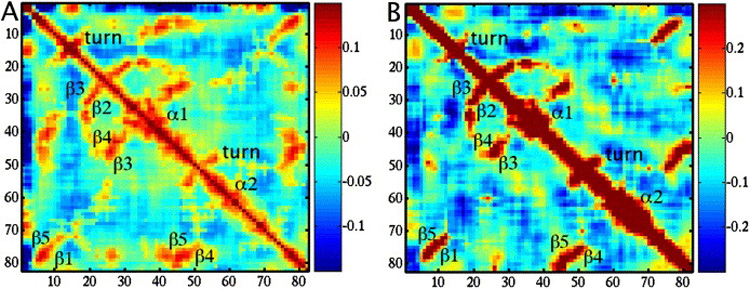
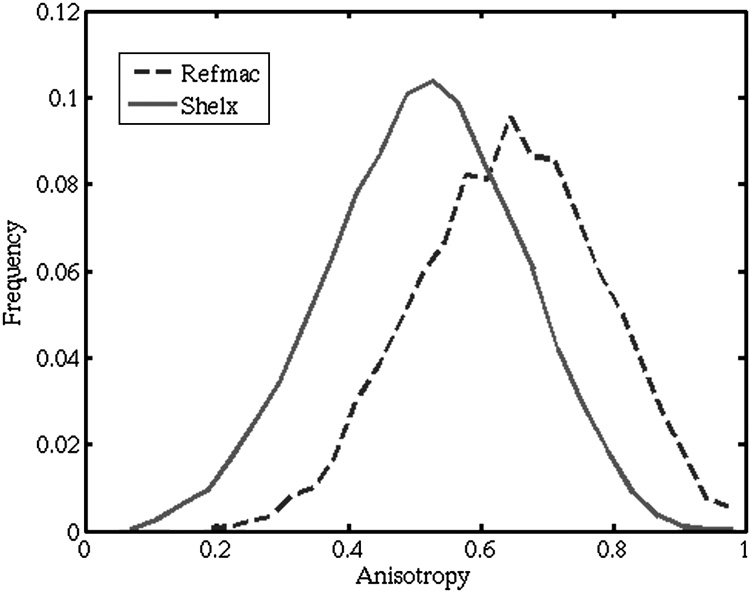
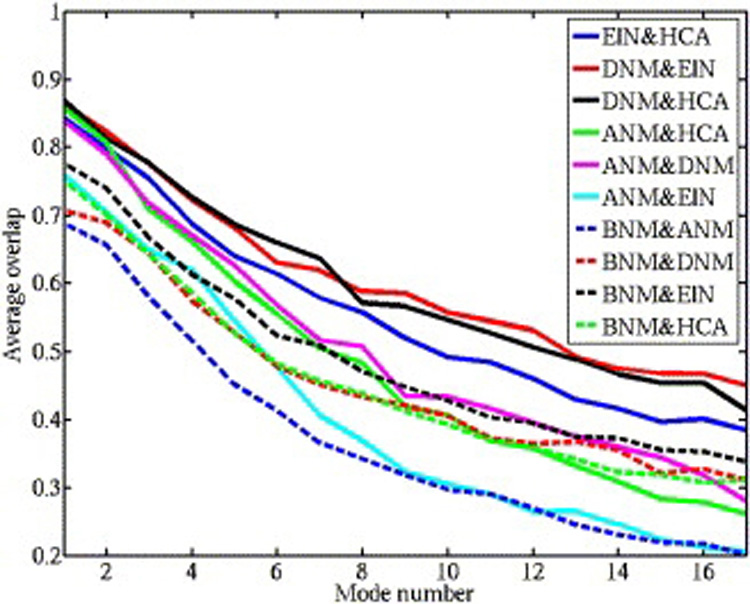
Kondrashov Dmitry A, Van Wynsberghe Adam W, Bannen Ryan M, Cui Qiang, Phillips George N
Department of Biochemistry, University of Wisconsin, Madison, Madison, WI 53706, USA.
Structure. 2007 Feb;15(2):169-77. doi: 10.1016/j.str.2006.12.006.
Normal mode analysis offers an efficient way of modeling the conformational flexibility of protein structures. We use anisotropic displacement parameters from crystallography to test the quality of prediction of both the magnitude and directionality of conformational flexibility. Normal modes from four simple elastic network model potentials and from the CHARMM force field are calculated for a data set of 83 diverse, ultrahigh-resolution crystal structures. While all five potentials provide good predictions of the magnitude of flexibility, all-atom potentials have a clear edge at prediction of directionality, and the CHARMM potential has the highest prediction quality. The low-frequency modes from different potentials are similar, but those computed from the CHARMM potential show the greatest difference from the elastic network models. The comprehensive evaluation demonstrates the costs and benefits of using normal mode potentials of varying complexity.
简正模式分析提供了一种对蛋白质结构的构象灵活性进行建模的有效方法。我们使用来自晶体学的各向异性位移参数来测试构象灵活性的大小和方向性预测的质量。针对83个不同的超高分辨率晶体结构数据集,计算了来自四种简单弹性网络模型势和CHARMM力场的简正模式。虽然所有五种势都能很好地预测灵活性的大小,但全原子势在方向性预测方面具有明显优势,且CHARMM势具有最高的预测质量。不同势的低频模式相似,但由CHARMM势计算得到的模式与弹性网络模型的差异最大。综合评估展示了使用不同复杂程度的简正模式势的成本和收益。