Rusch Douglas B, Halpern Aaron L, Sutton Granger, Heidelberg Karla B, Williamson Shannon, Yooseph Shibu, Wu Dongying, Eisen Jonathan A, Hoffman Jeff M, Remington Karin, Beeson Karen, Tran Bao, Smith Hamilton, Baden-Tillson Holly, Stewart Clare, Thorpe Joyce, Freeman Jason, Andrews-Pfannkoch Cynthia, Venter Joseph E, Li Kelvin, Kravitz Saul, Heidelberg John F, Utterback Terry, Rogers Yu-Hui, Falcón Luisa I, Souza Valeria, Bonilla-Rosso Germán, Eguiarte Luis E, Karl David M, Sathyendranath Shubha, Platt Trevor, Bermingham Eldredge, Gallardo Victor, Tamayo-Castillo Giselle, Ferrari Michael R, Strausberg Robert L, Nealson Kenneth, Friedman Robert, Frazier Marvin, Venter J Craig
J. Craig Venter Institute, Rockville, Maryland, United States of America.
PLoS Biol. 2007 Mar;5(3):e77. doi: 10.1371/journal.pbio.0050077.
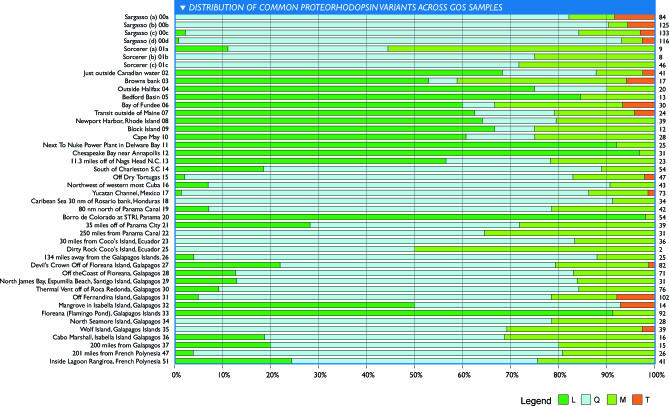
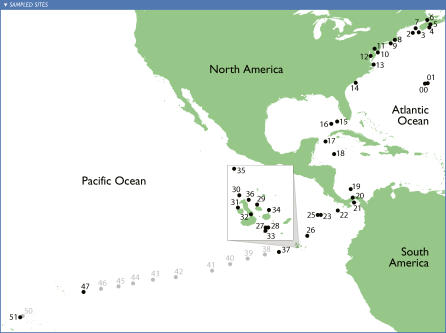
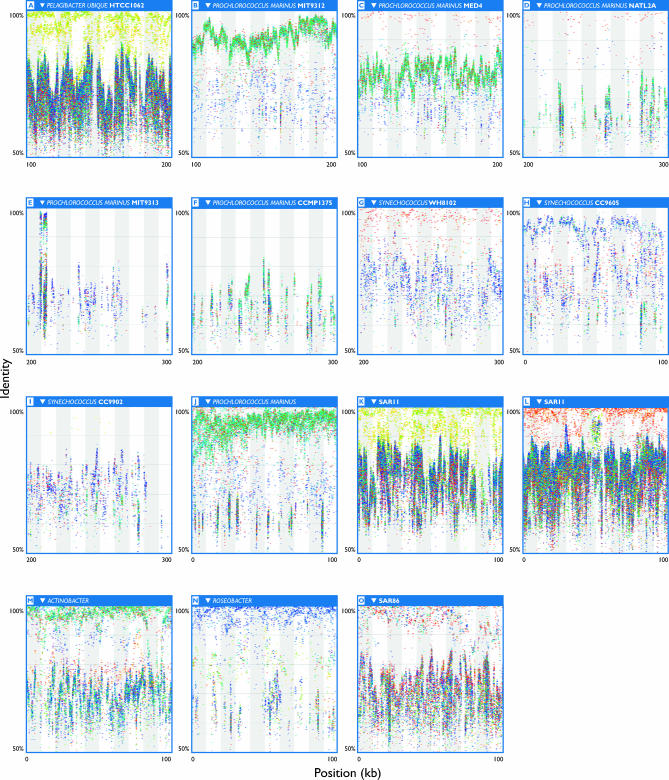
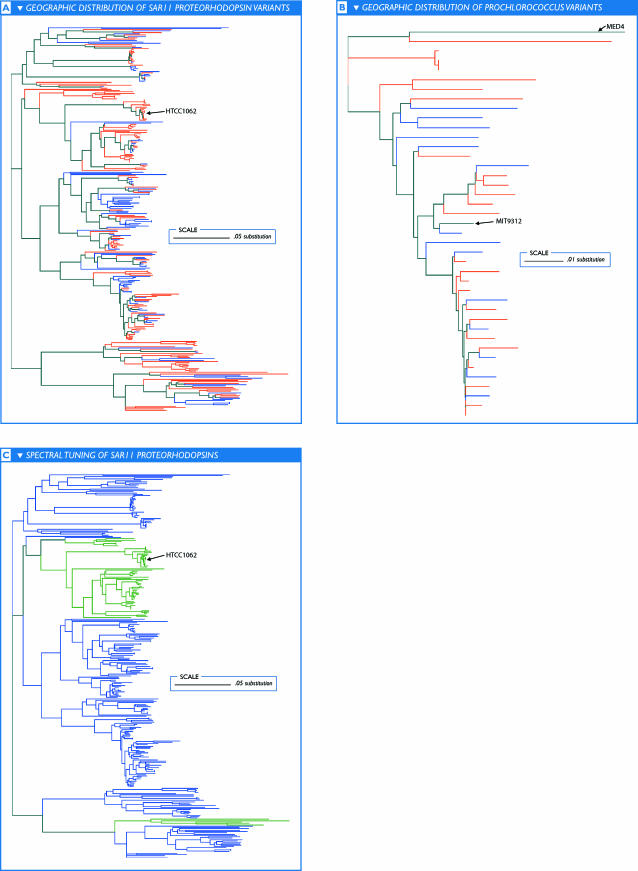
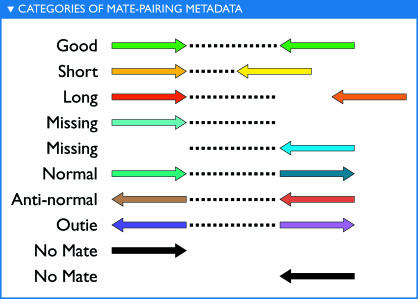
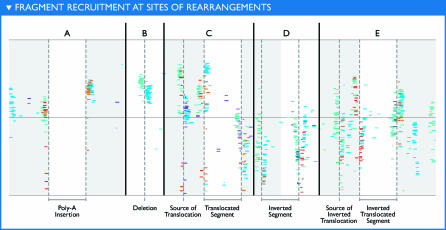
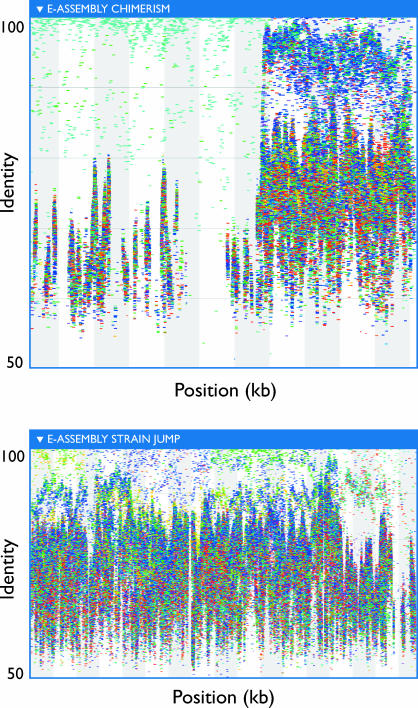
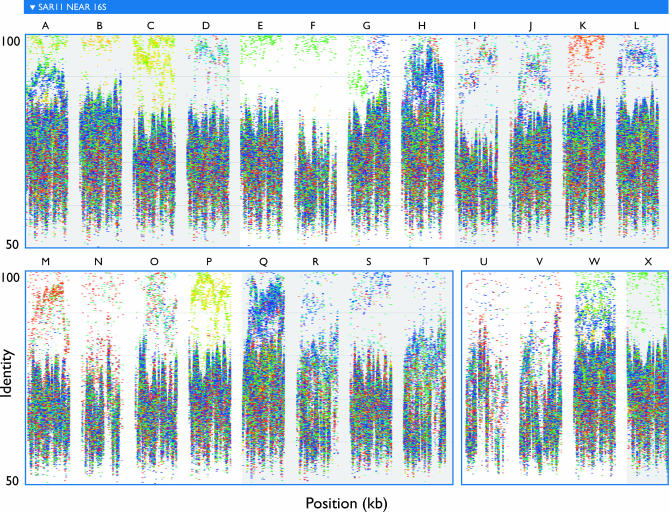
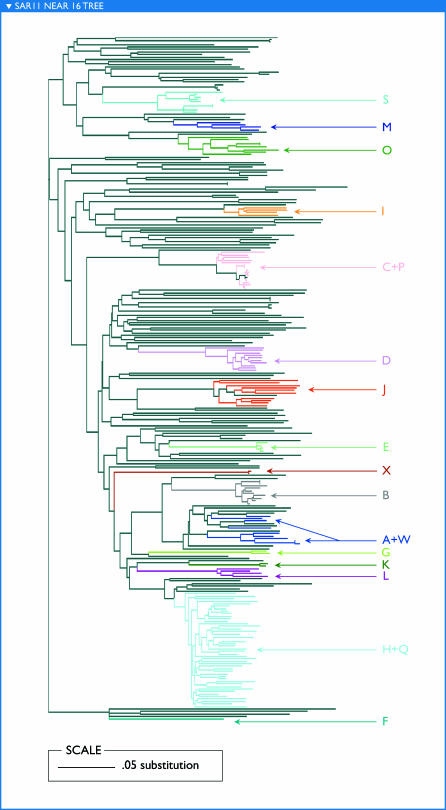
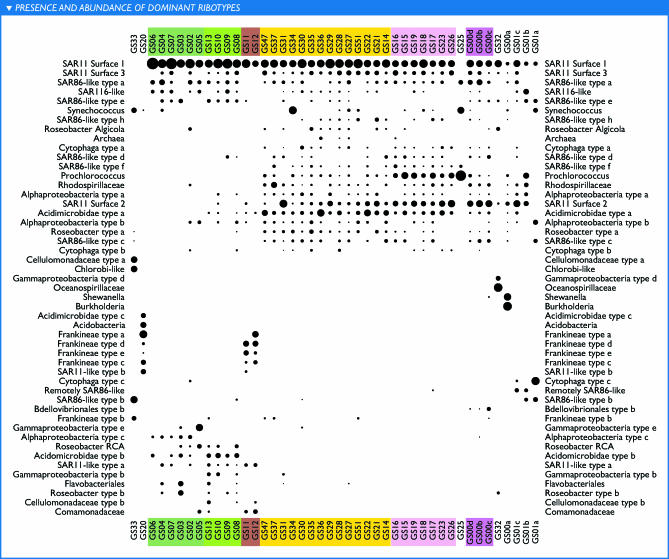
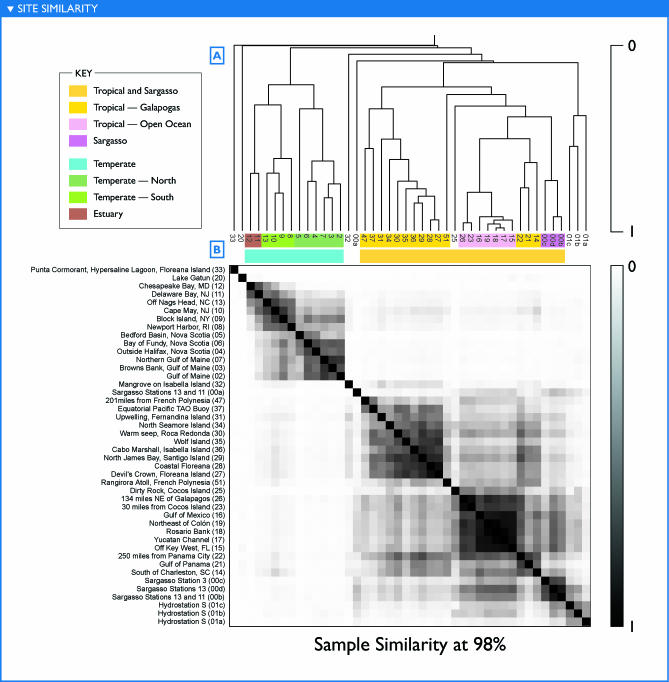
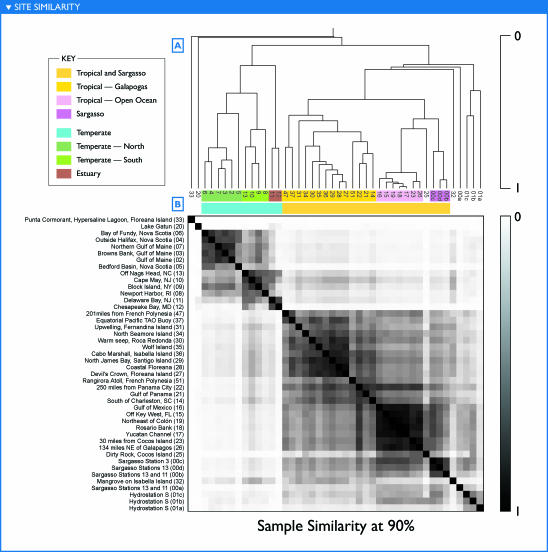
The world's oceans contain a complex mixture of micro-organisms that are for the most part, uncharacterized both genetically and biochemically. We report here a metagenomic study of the marine planktonic microbiota in which surface (mostly marine) water samples were analyzed as part of the Sorcerer II Global Ocean Sampling expedition. These samples, collected across a several-thousand km transect from the North Atlantic through the Panama Canal and ending in the South Pacific yielded an extensive dataset consisting of 7.7 million sequencing reads (6.3 billion bp). Though a few major microbial clades dominate the planktonic marine niche, the dataset contains great diversity with 85% of the assembled sequence and 57% of the unassembled data being unique at a 98% sequence identity cutoff. Using the metadata associated with each sample and sequencing library, we developed new comparative genomic and assembly methods. One comparative genomic method, termed "fragment recruitment," addressed questions of genome structure, evolution, and taxonomic or phylogenetic diversity, as well as the biochemical diversity of genes and gene families. A second method, termed "extreme assembly," made possible the assembly and reconstruction of large segments of abundant but clearly nonclonal organisms. Within all abundant populations analyzed, we found extensive intra-ribotype diversity in several forms: (1) extensive sequence variation within orthologous regions throughout a given genome; despite coverage of individual ribotypes approaching 500-fold, most individual sequencing reads are unique; (2) numerous changes in gene content some with direct adaptive implications; and (3) hypervariable genomic islands that are too variable to assemble. The intra-ribotype diversity is organized into genetically isolated populations that have overlapping but independent distributions, implying distinct environmental preference. We present novel methods for measuring the genomic similarity between metagenomic samples and show how they may be grouped into several community types. Specific functional adaptations can be identified both within individual ribotypes and across the entire community, including proteorhodopsin spectral tuning and the presence or absence of the phosphate-binding gene PstS.
世界海洋中含有复杂的微生物混合物,其中大部分在基因和生化方面都未得到表征。我们在此报告一项关于海洋浮游微生物群的宏基因组研究,在这项研究中,作为“魔法师二号”全球海洋采样探险的一部分,对表层(主要是海水)水样进行了分析。这些样本是从北大西洋经巴拿马运河至南太平洋,沿着数千公里的断面采集的,产生了一个由770万个测序读数(63亿碱基对)组成的庞大数据集。尽管少数主要的微生物类群在浮游海洋生态位中占主导地位,但该数据集包含了极大的多样性,在98%的序列同一性阈值下,85%的组装序列和57%的未组装数据是独特的。利用与每个样本和测序文库相关的元数据,我们开发了新的比较基因组学和组装方法。一种比较基因组学方法,称为“片段招募”,解决了基因组结构、进化、分类或系统发育多样性以及基因和基因家族的生化多样性问题。另一种方法,称为“极端组装”,使得组装和重建大量丰富但明显非克隆生物的大片段成为可能。在所有分析的丰富种群中,我们发现了几种形式的广泛的核糖型内多样性:(1)在给定基因组的直系同源区域内存在广泛的序列变异;尽管单个核糖型的覆盖度接近500倍,但大多数单个测序读数都是独特的;(2)基因含量有许多变化,其中一些具有直接的适应性影响;(3)高度可变的基因组岛,其变异性太大而无法组装。核糖型内多样性被组织成基因隔离的种群,这些种群具有重叠但独立的分布,这意味着不同的环境偏好。我们提出了测量宏基因组样本之间基因组相似性的新方法,并展示了如何将它们分组为几种群落类型。特定的功能适应性既可以在单个核糖型内识别,也可以在整个群落中识别,包括视紫质光谱调谐以及磷酸盐结合基因PstS的有无。