Mahony Shaun, Auron Philip E, Benos Panayiotis V
Department of Computational Biology, School of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America.
PLoS Comput Biol. 2007 Mar 30;3(3):e61. doi: 10.1371/journal.pcbi.0030061. Epub 2007 Feb 15.
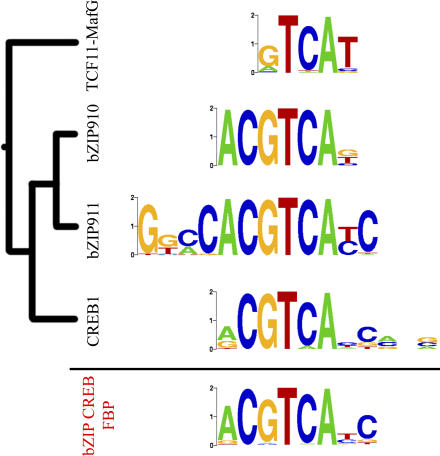
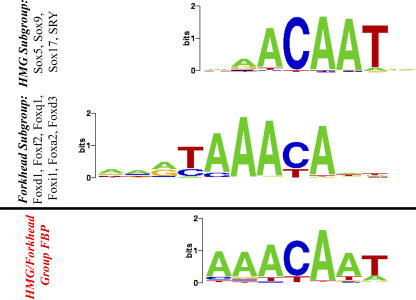
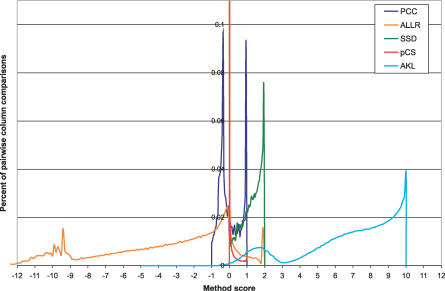
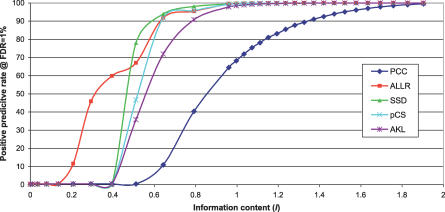
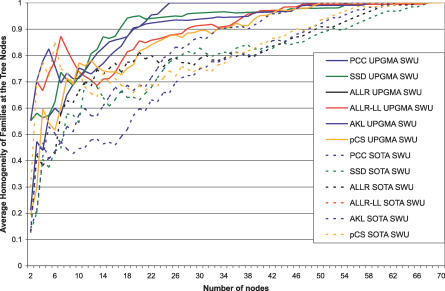
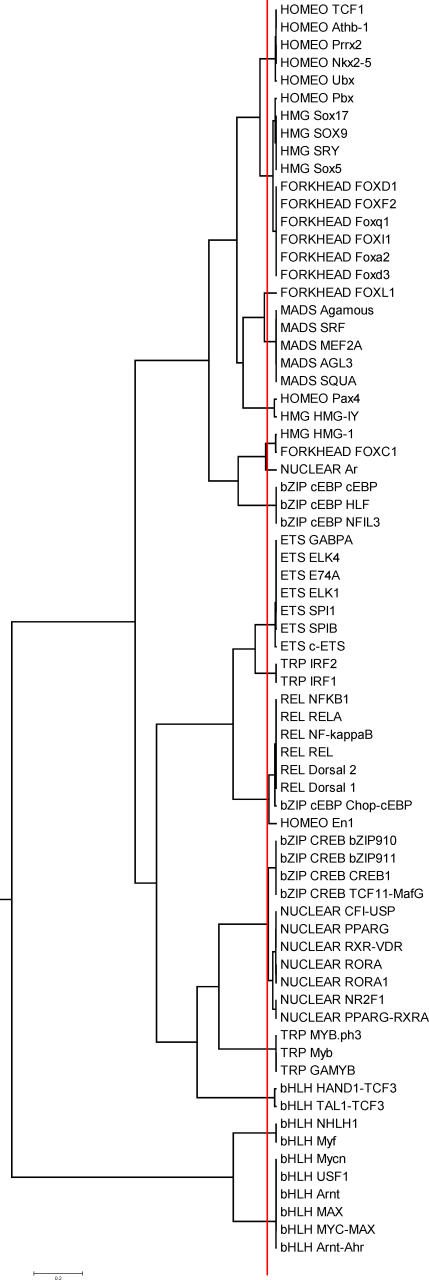
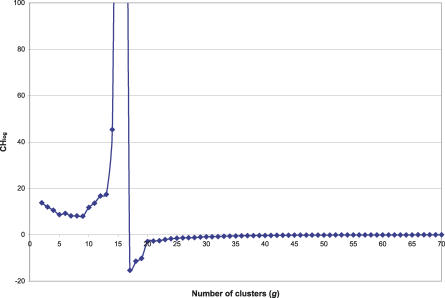
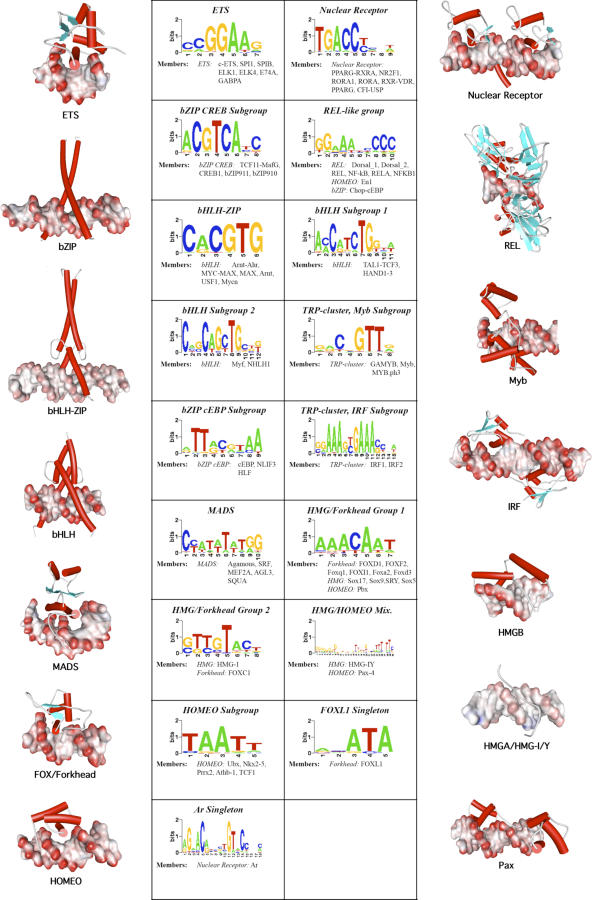
Transcription factor (TF) proteins recognize a small number of DNA sequences with high specificity and control the expression of neighbouring genes. The evolution of TF binding preference has been the subject of a number of recent studies, in which generalized binding profiles have been introduced and used to improve the prediction of new target sites. Generalized profiles are generated by aligning and merging the individual profiles of related TFs. However, the distance metrics and alignment algorithms used to compare the binding profiles have not yet been fully explored or optimized. As a result, binding profiles depend on TF structural information and sometimes may ignore important distinctions between subfamilies. Prediction of the identity or the structural class of a protein that binds to a given DNA pattern will enhance the analysis of microarray and ChIP-chip data where frequently multiple putative targets of usually unknown TFs are predicted. Various comparison metrics and alignment algorithms are evaluated (a total of 105 combinations). We find that local alignments are generally better than global alignments at detecting eukaryotic DNA motif similarities, especially when combined with the sum of squared distances or Pearson's correlation coefficient comparison metrics. In addition, multiple-alignment strategies for binding profiles and tree-building methods are tested for their efficiency in constructing generalized binding models. A new method for automatic determination of the optimal number of clusters is developed and applied in the construction of a new set of familial binding profiles which improves upon TF classification accuracy. A software tool, STAMP, is developed to host all tested methods and make them publicly available. This work provides a high quality reference set of familial binding profiles and the first comprehensive platform for analysis of DNA profiles. Detecting similarities between DNA motifs is a key step in the comparative study of transcriptional regulation, and the work presented here will form the basis for tool and method development for future transcriptional modeling studies.
转录因子(TF)蛋白能够高度特异性地识别少数DNA序列,并控制相邻基因的表达。TF结合偏好的进化一直是近期多项研究的主题,其中引入了广义结合谱并用于改进新靶位点的预测。广义谱是通过比对和合并相关TF的个体谱生成的。然而,用于比较结合谱的距离度量和比对算法尚未得到充分探索或优化。因此,结合谱依赖于TF结构信息,有时可能会忽略亚家族之间的重要差异。预测与给定DNA模式结合的蛋白质的身份或结构类别,将增强对微阵列和芯片数据的分析,在这些数据中,通常会预测多个通常未知TF的假定靶标。对各种比较度量和比对算法进行了评估(总共105种组合)。我们发现,在检测真核生物DNA基序相似性方面,局部比对通常优于全局比对,特别是当与平方距离之和或皮尔逊相关系数比较度量结合使用时。此外,还测试了结合谱的多重比对策略和建树方法在构建广义结合模型中的效率。开发了一种自动确定最佳聚类数的新方法,并将其应用于构建一组新的家族性结合谱,从而提高了TF分类的准确性。开发了一个软件工具STAMP,用于承载所有测试方法并使其公开可用。这项工作提供了一组高质量的家族性结合谱参考集以及第一个用于DNA谱分析的综合平台。检测DNA基序之间的相似性是转录调控比较研究中的关键步骤,本文介绍的工作将为未来转录建模研究的工具和方法开发奠定基础。