Harmanci Arif Ozgun, Sharma Gaurav, Mathews David H
Department of Electrical and Computer Engineering, University of Rochester, Hopeman 204, Rochester, NY 14627, USA.
BMC Bioinformatics. 2007 Apr 19;8:130. doi: 10.1186/1471-2105-8-130.
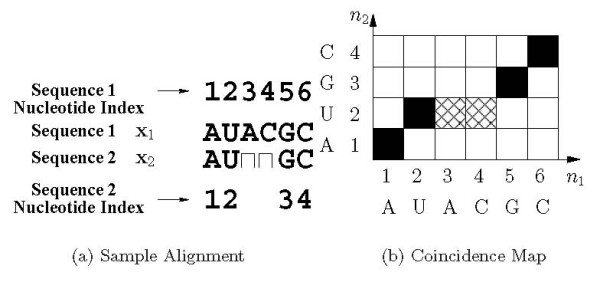
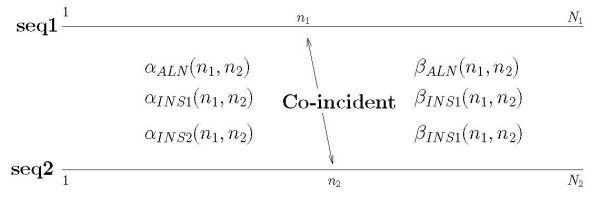
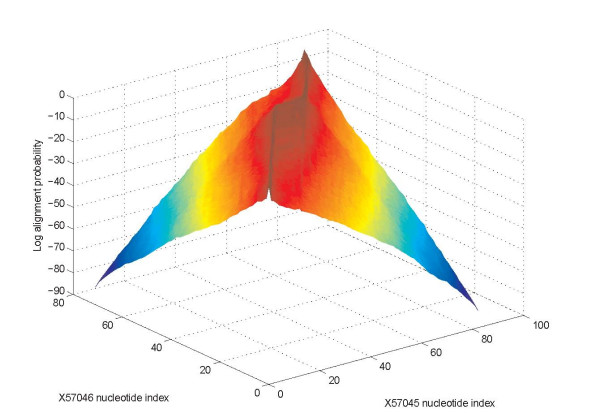
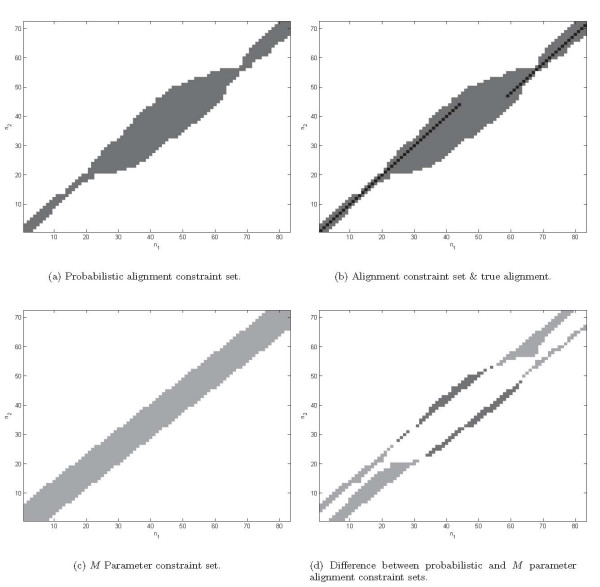
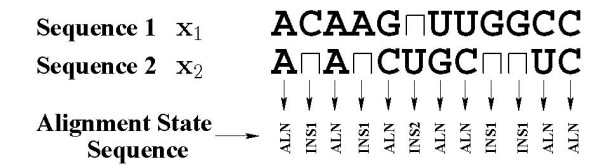
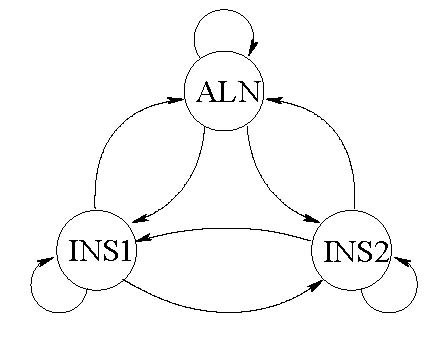
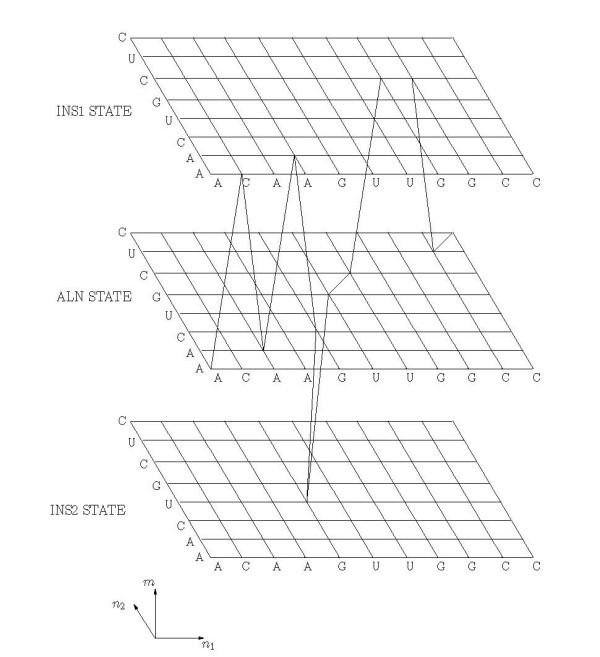
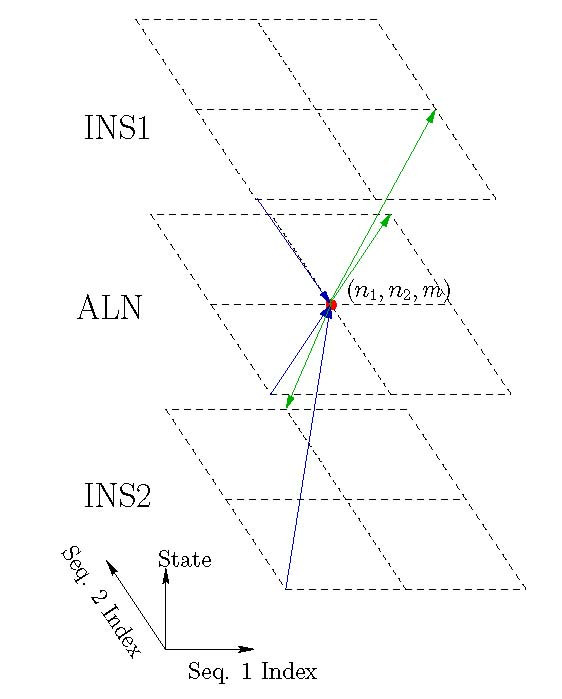
Joint alignment and secondary structure prediction of two RNA sequences can significantly improve the accuracy of the structural predictions. Methods addressing this problem, however, are forced to employ constraints that reduce computation by restricting the alignments and/or structures (i.e. folds) that are permissible. In this paper, a new methodology is presented for the purpose of establishing alignment constraints based on nucleotide alignment and insertion posterior probabilities. Using a hidden Markov model, posterior probabilities of alignment and insertion are computed for all possible pairings of nucleotide positions from the two sequences. These alignment and insertion posterior probabilities are additively combined to obtain probabilities of co-incidence for nucleotide position pairs. A suitable alignment constraint is obtained by thresholding the co-incidence probabilities. The constraint is integrated with Dynalign, a free energy minimization algorithm for joint alignment and secondary structure prediction. The resulting method is benchmarked against the previous version of Dynalign and against other programs for pairwise RNA structure prediction.
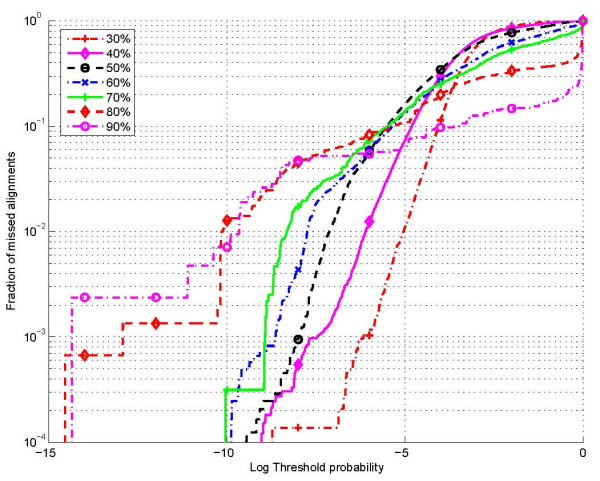
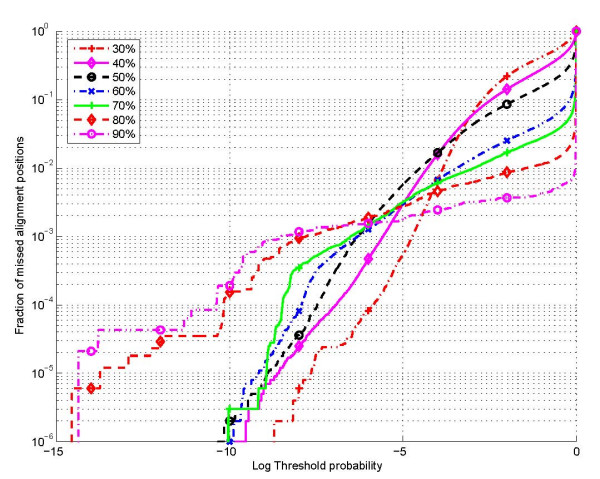
The proposed technique eliminates manual parameter selection in Dynalign and provides significant computational time savings in comparison to prior constraints in Dynalign while simultaneously providing a small improvement in the structural prediction accuracy. Savings are also realized in memory. In experiments over a 5S RNA dataset with average sequence length of approximately 120 nucleotides, the method reduces computation by a factor of 2. The method performs favorably in comparison to other programs for pairwise RNA structure prediction: yielding better accuracy, on average, and requiring significantly lesser computational resources.
Probabilistic analysis can be utilized in order to automate the determination of alignment constraints for pairwise RNA structure prediction methods in a principled fashion. These constraints can reduce the computational and memory requirements of these methods while maintaining or improving their accuracy of structural prediction. This extends the practical reach of these methods to longer length sequences. The revised Dynalign code is freely available for download.
两条RNA序列的联合比对和二级结构预测能够显著提高结构预测的准确性。然而,解决此问题的方法不得不采用一些约束条件,通过限制允许的比对和/或结构(即折叠)来减少计算量。本文提出了一种新方法,旨在基于核苷酸比对和插入后验概率来建立比对约束。使用隐马尔可夫模型,计算两条序列中所有可能核苷酸位置对的比对和插入后验概率。将这些比对和插入后验概率相加组合,以获得核苷酸位置对的重合概率。通过对比对重合概率进行阈值化处理,得到合适的比对约束。该约束与Dynalign(一种用于联合比对和二级结构预测的自由能最小化算法)相结合。将所得方法与Dynalign的先前版本以及其他用于成对RNA结构预测的程序进行基准测试。
所提出的技术消除了Dynalign中的手动参数选择,与Dynalign先前的约束相比,显著节省了计算时间,同时在结构预测准确性上有小幅提高。在内存方面也实现了节省。在一个平均序列长度约为120个核苷酸的5S RNA数据集上进行的实验中,该方法将计算量减少了一半。与其他用于成对RNA结构预测的程序相比,该方法表现良好:平均而言,具有更高的准确性,并且所需的计算资源显著更少。
可以利用概率分析以一种有原则的方式自动确定成对RNA结构预测方法的比对约束。这些约束可以降低这些方法的计算和内存需求,同时保持或提高其结构预测的准确性。这将这些方法的实际应用范围扩展到了更长的序列。修订后的Dynalign代码可免费下载。