Shmygelska Alena, Hoos Holger H
Department of Structural Biology, Stanford University, Stanford, CA 94305, USA.
BMC Bioinformatics. 2007 Apr 24;8:136. doi: 10.1186/1471-2105-8-136.
The problem of protein structure prediction consists of predicting the functional or native structure of a protein given its linear sequence of amino acids. This problem has played a prominent role in the fields of biomolecular physics and algorithm design for over 50 years. Additionally, its importance increases continually as a result of an exponential growth over time in the number of known protein sequences in contrast to a linear increase in the number of determined structures. Our work focuses on the problem of searching an exponentially large space of possible conformations as efficiently as possible, with the goal of finding a global optimum with respect to a given energy function. This problem plays an important role in the analysis of systems with complex search landscapes, and particularly in the context of ab initio protein structure prediction.
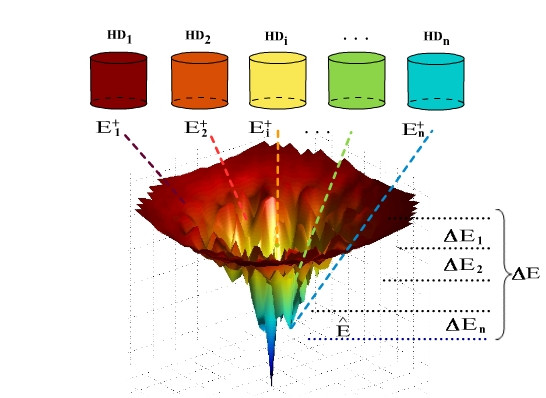
In this work, we introduce a novel approach for solving this conformation search problem based on the use of a bin framework for adaptively storing and retrieving promising locally optimal solutions. Our approach provides a rich and general framework within which a broad range of adaptive or reactive search strategies can be realized. Here, we introduce adaptive mechanisms for choosing which conformations should be stored, based on the set of conformations already stored in memory, and for biasing choices when retrieving conformations from memory in order to overcome search stagnation.
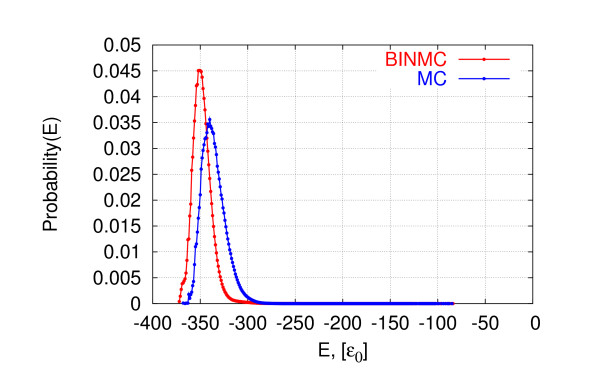
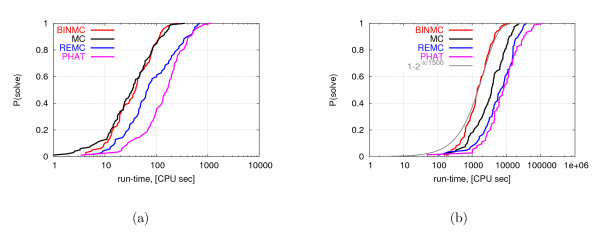
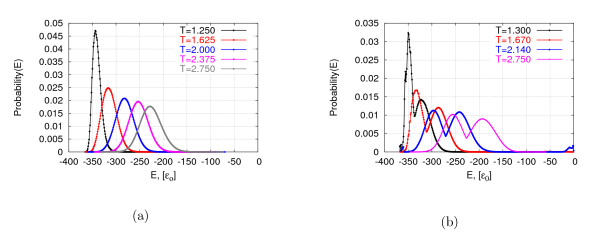
We show that our bin framework combined with a widely used optimization method, Monte Carlo search, achieves significantly better performance than state-of-the-art generalized ensemble methods for a well-known protein-like homopolymer model on the face-centered cubic lattice.
蛋白质结构预测问题包括根据蛋白质的氨基酸线性序列预测其功能结构或天然结构。在过去50多年里,这个问题在生物分子物理学和算法设计领域一直扮演着重要角色。此外,由于已知蛋白质序列数量随时间呈指数增长,而已确定结构的数量呈线性增长,其重要性也在不断增加。我们的工作重点是尽可能高效地搜索指数级庞大的可能构象空间,目标是找到相对于给定能量函数的全局最优解。这个问题在具有复杂搜索格局的系统分析中,特别是在从头开始的蛋白质结构预测背景下,起着重要作用。
在这项工作中,我们引入了一种新颖的方法来解决这个构象搜索问题,该方法基于使用一个箱式框架来自适应地存储和检索有前景的局部最优解。我们的方法提供了一个丰富且通用的框架,在其中可以实现广泛的自适应或反应式搜索策略。在这里,我们引入了自适应机制,用于根据已存储在内存中的构象集选择应存储哪些构象,以及在从内存中检索构象时偏向选择以克服搜索停滞。
我们表明,对于面心立方晶格上的一个著名的类蛋白质均聚物模型,我们的箱式框架与广泛使用的优化方法蒙特卡罗搜索相结合,比现有最先进的广义系综方法具有显著更好的性能。