Song Robert X-D, Zhang Zhenguo, Chen Yucai, Bao Yongde, Santen Richard J
Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, Virginia 22903, USA.
Endocrinology. 2007 Aug;148(8):4091-101. doi: 10.1210/en.2007-0240. Epub 2007 May 24.
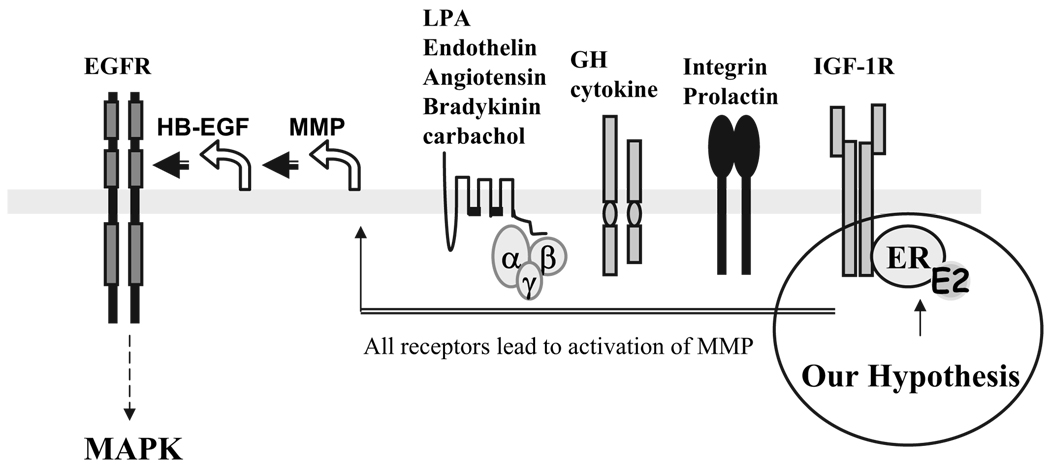
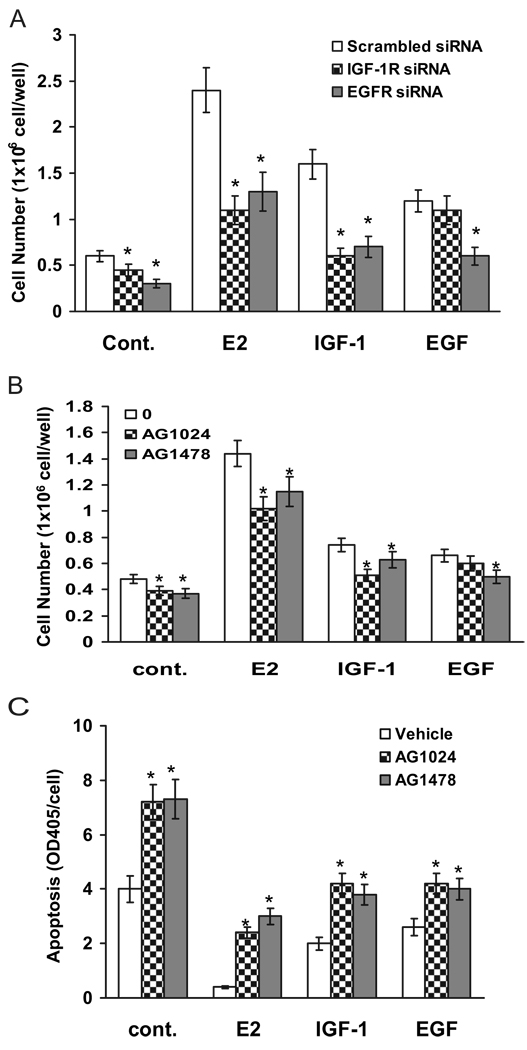
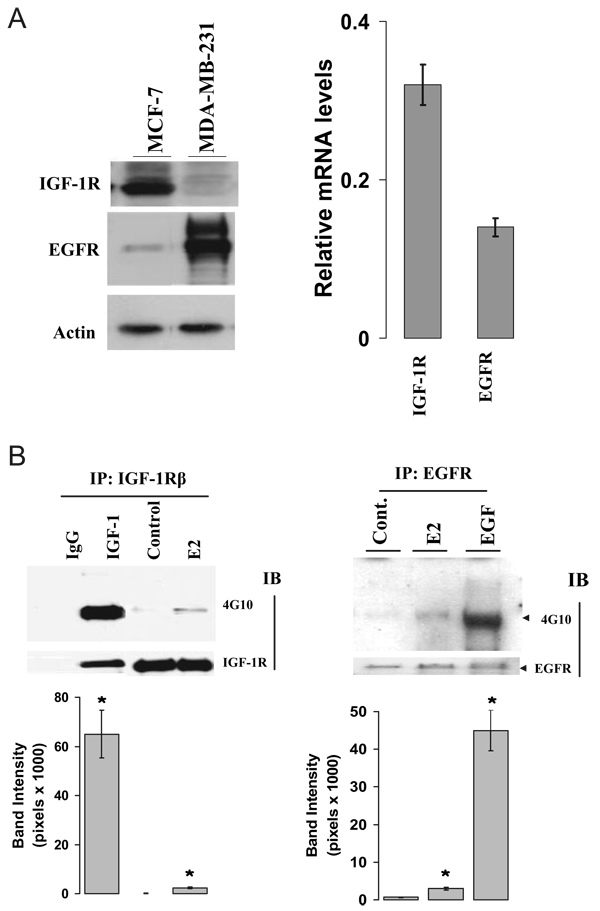
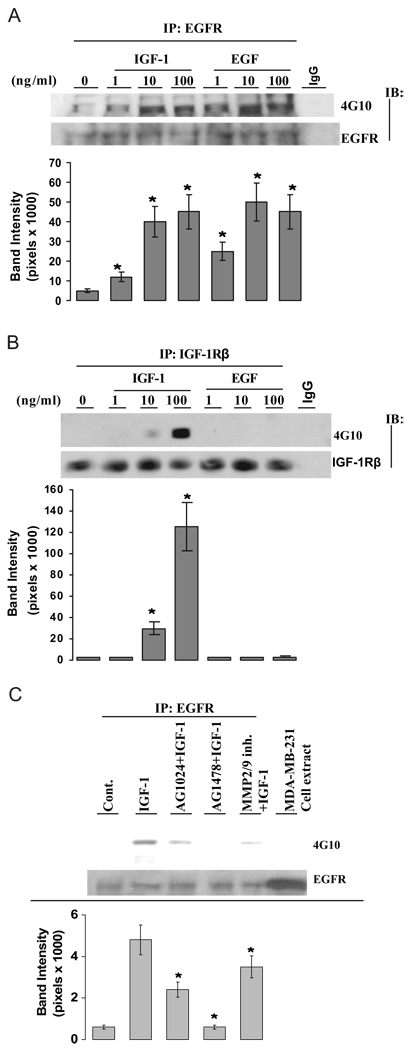
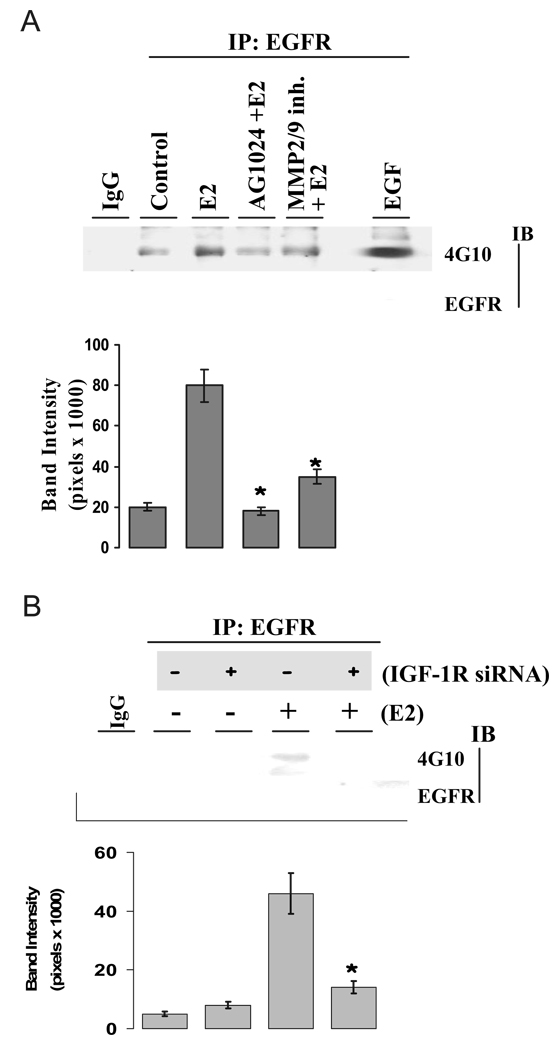
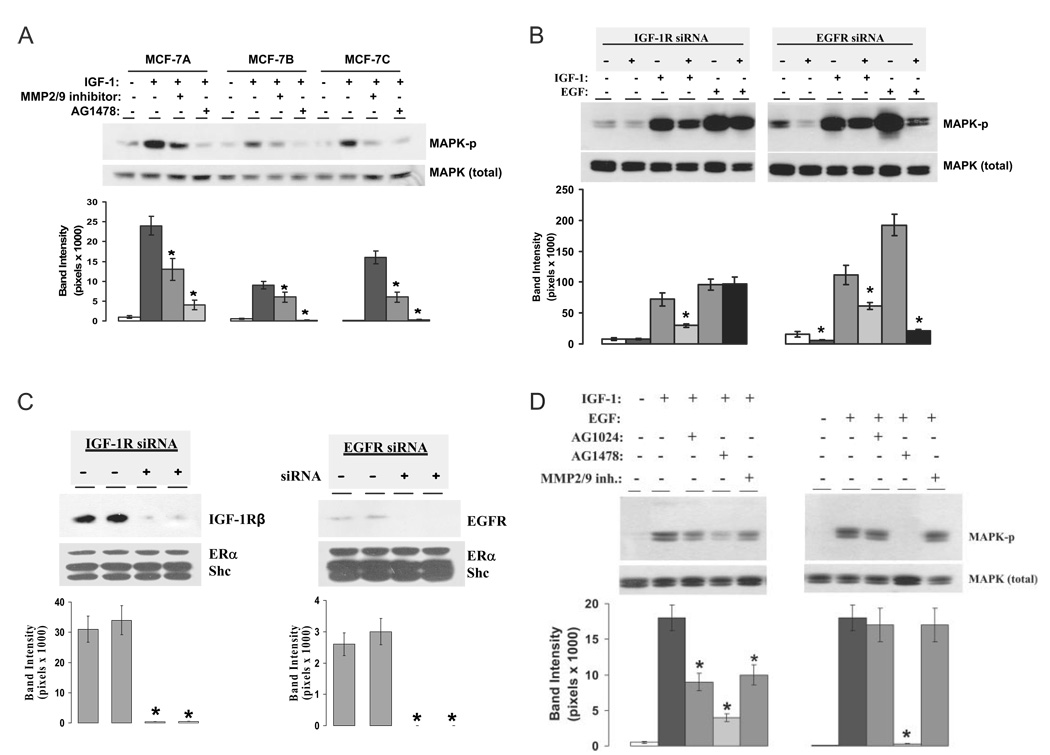
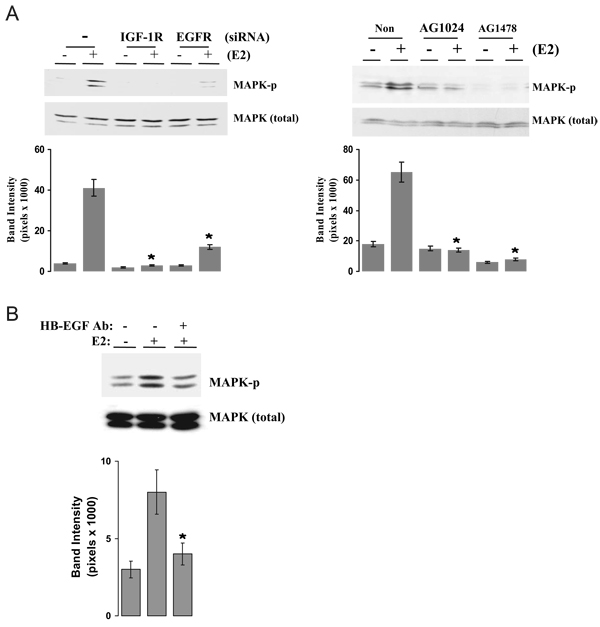
We present an integrated model of an extranuclear, estrogen receptor-alpha (ERalpha)-mediated, rapid MAPK activation pathway in breast cancer cells. In noncancer cells, IGF-I initiates a linear process involving activation of the IGF-I receptor (IGF-IR) and matrix metalloproteinases (MMP), release of heparin-binding epidermal growth factor (HB-EGF), and activation of EGF receptor (EGFR)-dependent MAPK. 17beta-Estradiol (E2) rapidly activates IGF-IR in breast cancer cells. We hypothesize that E2 induces a similar linear pathway involving IGF-IR, MMP, HB-EGF, EGFR, and MAPK. Using MCF-7 breast cancer cells, we for the first time demonstrated that a sequential activation of IGF-IR, MMP, and EGFR existed in E2 and IGF-I actions, which was supported by evidence that the selective inhibitors of IGF-IR and MMP or knockdown of IGF-IR all inhibited E2- or IGF-I-induced EGFR phosphorylation. Using the inhibitors and small inhibitory RNA strategies, we also demonstrated that the same sequential activation of the receptors occurred in E2-, IGF-I-, but not EGF-induced MAPK phosphorylation. Additionally, a HB-EGF neutralizing antibody significantly blocked E2-induced MAPK activation, further supporting our hypothesis. The biological effects of sequential activation of IGF-IR and EGFR on E2 stimulation of cell proliferation were also investigated. Knockdown or blockade of IGF-IR significantly inhibited E2- or IGF-I-stimulated but not EGF-induced cell growth. Knockdown or blockade of EGFR abrogated cell growth induced by E2, IGF-I, and EGF, indicating that EGFR is a downstream molecule of IGF-IR in E2 and IGF-I action. Together, our data support the novel view that E2 can activate a linear pathway involving the sequential activation of IGF-IR, MMP, HB-EGF, EGFR, and MAPK.
我们提出了一种乳腺癌细胞中细胞核外雌激素受体α(ERα)介导的快速丝裂原活化蛋白激酶(MAPK)激活途径的整合模型。在非癌细胞中,胰岛素样生长因子I(IGF-I)启动一个线性过程,涉及胰岛素样生长因子I受体(IGF-IR)和基质金属蛋白酶(MMP)的激活、肝素结合表皮生长因子(HB-EGF)的释放以及表皮生长因子受体(EGFR)依赖性MAPK的激活。17β-雌二醇(E2)可快速激活乳腺癌细胞中的IGF-IR。我们推测E2诱导了一条涉及IGF-IR、MMP、HB-EGF、EGFR和MAPK的相似线性途径。使用MCF-7乳腺癌细胞,我们首次证明在E2和IGF-I作用中存在IGF-IR、MMP和EGFR的顺序激活,这一观点得到如下证据支持:IGF-IR和MMP的选择性抑制剂或IGF-IR的敲低均抑制E2或IGF-I诱导的EGFR磷酸化。使用抑制剂和小干扰RNA策略,我们还证明在E2、IGF-I而非EGF诱导的MAPK磷酸化过程中发生了相同的受体顺序激活。此外,一种HB-EGF中和抗体显著阻断E2诱导的MAPK激活,进一步支持了我们的推测。我们还研究了IGF-IR和EGFR顺序激活对E2刺激细胞增殖的生物学效应。IGF-IR的敲低或阻断显著抑制E2或IGF-I刺激的细胞生长,但不抑制EGF诱导的细胞生长。EGFR的敲低或阻断消除了E2、IGF-I和EGF诱导的细胞生长,表明在E2和IGF-I作用中EGFR是IGF-IR的下游分子。总之,我们的数据支持一种新观点,即E2可激活一条涉及IGF-IR、MMP、HB-EGF、EGFR和MAPK顺序激活的线性途径。