Macconaill Laura E, Aldred Micheala A, Lu Xincheng, Laframboise Thomas
Dana-Farber Cancer Institute, Boston, Massachusetts 02116, USA.
BMC Genomics. 2007 Jul 3;8:211. doi: 10.1186/1471-2164-8-211.
The recent discovery of widespread copy number variation in humans has forced a shift away from the assumption of two copies per locus per cell throughout the autosomal genome. In particular, a SNP site can no longer always be accurately assigned one of three genotypes in an individual. In the presence of copy number variability, the individual may theoretically harbor any number of copies of each of the two SNP alleles.
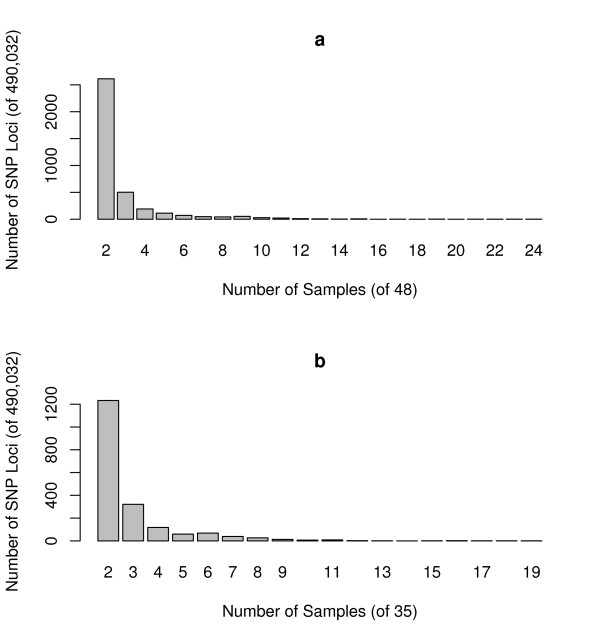
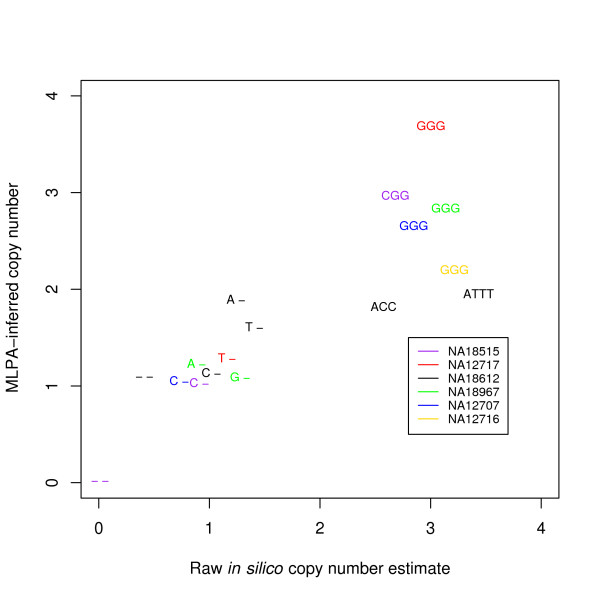
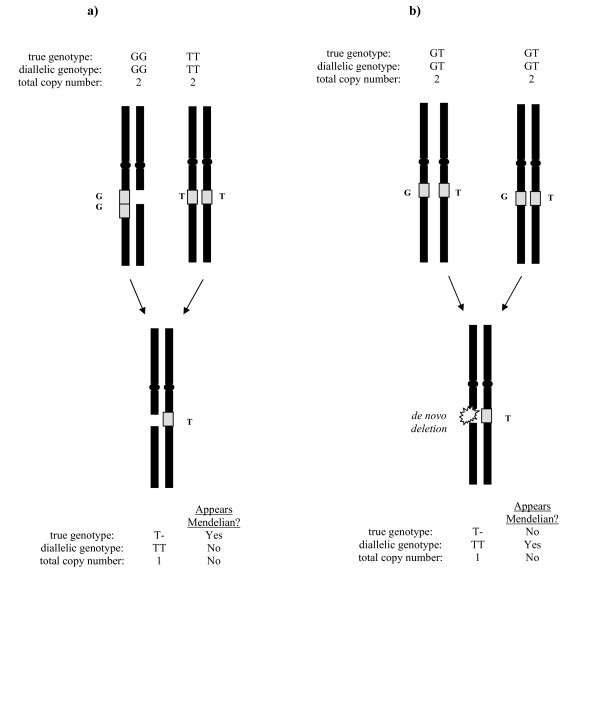
To address this issue, we have developed a method to infer a "generalized genotype" from raw SNP microarray data. Here we apply our approach to data from 48 individuals and uncover thousands of aberrant SNPs, most in regions that were previously unreported as copy number variants. We show that our allele-specific copy numbers follow Mendelian inheritance patterns that would be obscured in the absence of SNP allele information. The interplay between duplication and point mutation in our data shed light on the relative frequencies of these events in human history, showing that at least some of the duplication events were recurrent.
This new multi-allelic view of SNPs has a complicated role in disease association studies, and further work will be necessary in order to accurately assess its importance. Software to perform generalized genotyping from SNP array data is freely available online 1.
近期在人类中发现广泛存在的拷贝数变异,迫使人们摒弃了常染色体基因组中每个位点每个细胞有两个拷贝的假设。特别是,在个体中,一个单核苷酸多态性(SNP)位点不再总能准确地被指定为三种基因型之一。在存在拷贝数变异的情况下,理论上个体可能携带两个SNP等位基因中每个等位基因的任意数量的拷贝。
为解决这个问题,我们开发了一种从原始SNP微阵列数据推断“广义基因型”的方法。在此,我们将我们的方法应用于48个个体的数据,并发现了数千个异常SNP,其中大多数位于以前未报告为拷贝数变异的区域。我们表明,我们的等位基因特异性拷贝数遵循孟德尔遗传模式,而在没有SNP等位基因信息的情况下这些模式会被掩盖。我们数据中重复与点突变之间的相互作用揭示了这些事件在人类历史中的相对频率,表明至少一些重复事件是反复发生的。
这种关于SNP的新的多等位基因观点在疾病关联研究中具有复杂的作用,为了准确评估其重要性,还需要进一步开展工作。可从网上免费获取用于从SNP阵列数据进行广义基因分型的软件1。