Huse Susan M, Huber Julie A, Morrison Hilary G, Sogin Mitchell L, Welch David Mark
Josephine Bay Paul Center, Marine Biological Laboratory at Woods Hole, MBL Street, Woods Hole, MA 02543, USA.
Genome Biol. 2007;8(7):R143. doi: 10.1186/gb-2007-8-7-r143.
Massively parallel pyrosequencing systems have increased the efficiency of DNA sequencing, although the published per-base accuracy of a Roche GS20 is only 96%. In genome projects, highly redundant consensus assemblies can compensate for sequencing errors. In contrast, studies of microbial diversity that catalogue differences between PCR amplicons of ribosomal RNA genes (rDNA) or other conserved gene families cannot take advantage of consensus assemblies to detect and minimize incorrect base calls.
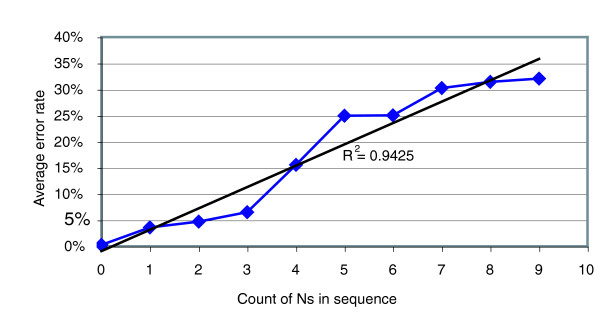
We performed an empirical study of the per-base error rate for the Roche GS20 system using sequences of the V6 hypervariable region from cloned microbial ribosomal DNA (tag sequencing). We calculated a 99.5% accuracy rate in unassembled sequences, and identified several factors that can be used to remove a small percentage of low-quality reads, improving the accuracy to 99.75% or better.
By using objective criteria to eliminate low quality data, the quality of individual GS20 sequence reads in molecular ecological applications can surpass the accuracy of traditional capillary methods.
大规模平行焦磷酸测序系统提高了DNA测序的效率,尽管罗氏GS20公布的每碱基准确率仅为96%。在基因组计划中,高度冗余的一致性组装可以弥补测序错误。相比之下,对核糖体RNA基因(rDNA)或其他保守基因家族的PCR扩增子之间差异进行编目的微生物多样性研究,无法利用一致性组装来检测并尽量减少错误的碱基识别。
我们使用来自克隆的微生物核糖体DNA的V6高变区序列(标签测序),对罗氏GS20系统的每碱基错误率进行了实证研究。我们计算出未组装序列的准确率为99.5%,并确定了几个可用于去除一小部分低质量读数的因素,将准确率提高到99.75%或更高。
通过使用客观标准消除低质量数据,在分子生态学应用中,单个GS20序列读数的质量可以超过传统毛细管方法的准确率。