Zotenko Elena, Dogan Rezarta Islamaj, Wilbur W John, O'Leary Dianne P, Przytycka Teresa M
Department of Computer Science, University of Maryland, College Park, MD 20742, USA.
BMC Struct Biol. 2007 Aug 9;7:53. doi: 10.1186/1472-6807-7-53.
One approach for speeding-up protein structure comparison is the projection approach, where a protein structure is mapped to a high-dimensional vector and structural similarity is approximated by distance between the corresponding vectors. Structural footprinting methods are projection methods that employ the same general technique to produce the mapping: first select a representative set of structural fragments as models and then map a protein structure to a vector in which each dimension corresponds to a particular model and "counts" the number of times the model appears in the structure. The main difference between any two structural footprinting methods is in the set of models they use; in fact a large number of methods can be generated by varying the type of structural fragments used and the amount of detail in their representation. How do these choices affect the ability of the method to detect various types of structural similarity?
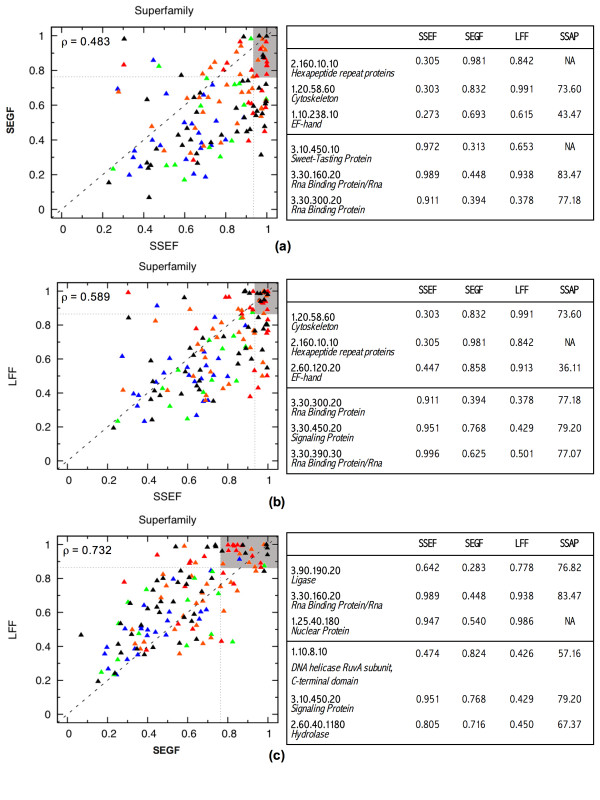
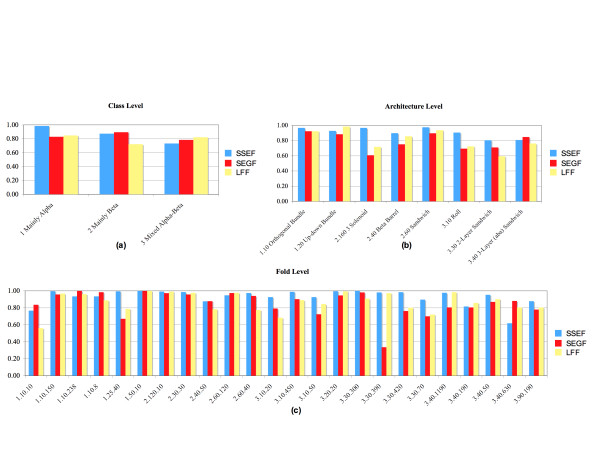
To answer this question we benchmarked three structural footprinting methods that vary significantly in their selection of models against the CATH database. In the first set of experiments we compared the methods' ability to detect structural similarity characteristic of evolutionarily related structures, i.e., structures within the same CATH superfamily. In the second set of experiments we tested the methods' agreement with the boundaries imposed by classification groups at the Class, Architecture, and Fold levels of the CATH hierarchy.
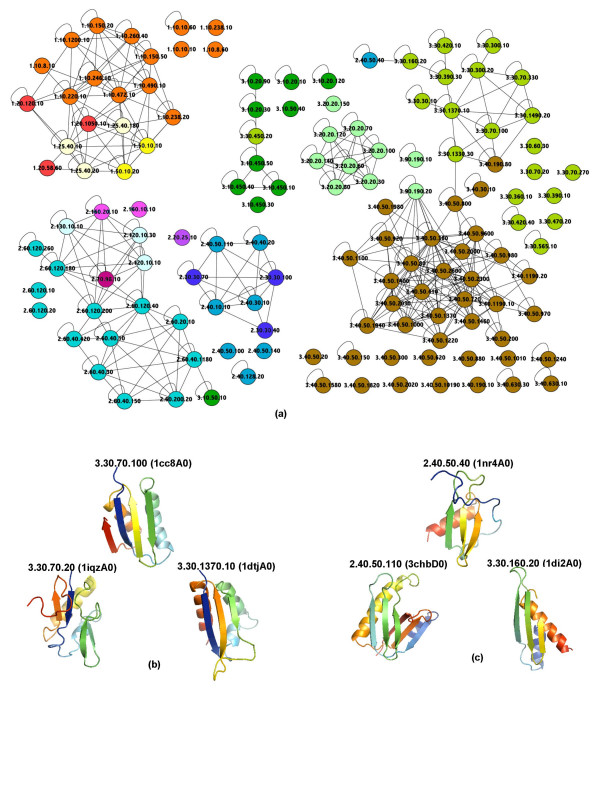
In both experiments we found that the method which uses secondary structure information has the best performance on average, but no one method performs consistently the best across all groups at a given classification level. We also found that combining the methods' outputs significantly improves the performance. Moreover, our new techniques to measure and visualize the methods' agreement with the CATH hierarchy, including the threshholded affinity graph, are useful beyond this work. In particular, they can be used to expose a similar composition of different classification groups in terms of structural fragments used by the method and thus provide an alternative demonstration of the continuous nature of the protein structure universe.
加速蛋白质结构比较的一种方法是投影法,即将蛋白质结构映射到高维向量,并通过相应向量之间的距离来近似结构相似性。结构足迹法是采用相同通用技术进行映射的投影法:首先选择一组具有代表性的结构片段作为模型,然后将蛋白质结构映射到一个向量中,其中每个维度对应一个特定模型,并“计算”该模型在结构中出现的次数。任何两种结构足迹法之间的主要区别在于它们所使用的模型集;实际上,通过改变所使用的结构片段类型及其表示中的细节量,可以生成大量方法。这些选择如何影响该方法检测各种类型结构相似性的能力?
为了回答这个问题,我们针对CATH数据库对三种在模型选择上有显著差异的结构足迹法进行了基准测试。在第一组实验中,我们比较了这些方法检测进化相关结构(即同一CATH超家族内的结构)所特有的结构相似性的能力。在第二组实验中,我们测试了这些方法与CATH层次结构的类、架构和折叠级别上分类组所划定边界的一致性。
在这两组实验中,我们发现使用二级结构信息的方法平均表现最佳,但在给定分类级别下,没有一种方法在所有组中始终表现最佳。我们还发现,将这些方法的输出结果相结合可显著提高性能。此外,我们用于测量和可视化这些方法与CATH层次结构一致性的新技术,包括阈值亲和图,在这项工作之外也很有用。特别是,它们可用于揭示不同分类组在方法所使用的结构片段方面的相似组成,从而为蛋白质结构宇宙的连续性提供另一种证明。