Luo Feng, Yang Yunfeng, Zhong Jianxin, Gao Haichun, Khan Latifur, Thompson Dorothea K, Zhou Jizhong
Environmental Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA.
BMC Bioinformatics. 2007 Aug 14;8:299. doi: 10.1186/1471-2105-8-299.
Large-scale sequencing of entire genomes has ushered in a new age in biology. One of the next grand challenges is to dissect the cellular networks consisting of many individual functional modules. Defining co-expression networks without ambiguity based on genome-wide microarray data is difficult and current methods are not robust and consistent with different data sets. This is particularly problematic for little understood organisms since not much existing biological knowledge can be exploited for determining the threshold to differentiate true correlation from random noise. Random matrix theory (RMT), which has been widely and successfully used in physics, is a powerful approach to distinguish system-specific, non-random properties embedded in complex systems from random noise. Here, we have hypothesized that the universal predictions of RMT are also applicable to biological systems and the correlation threshold can be determined by characterizing the correlation matrix of microarray profiles using random matrix theory.
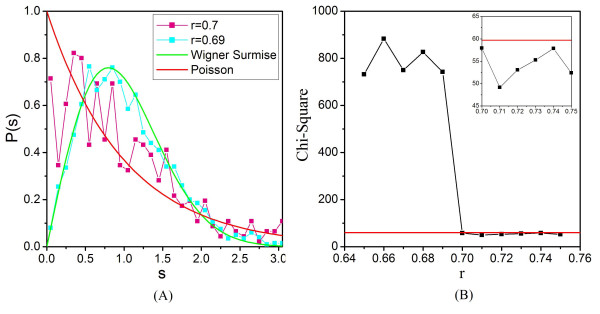
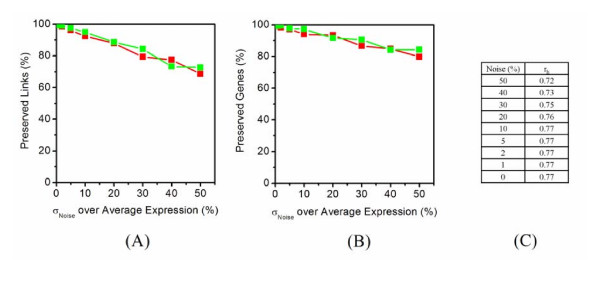
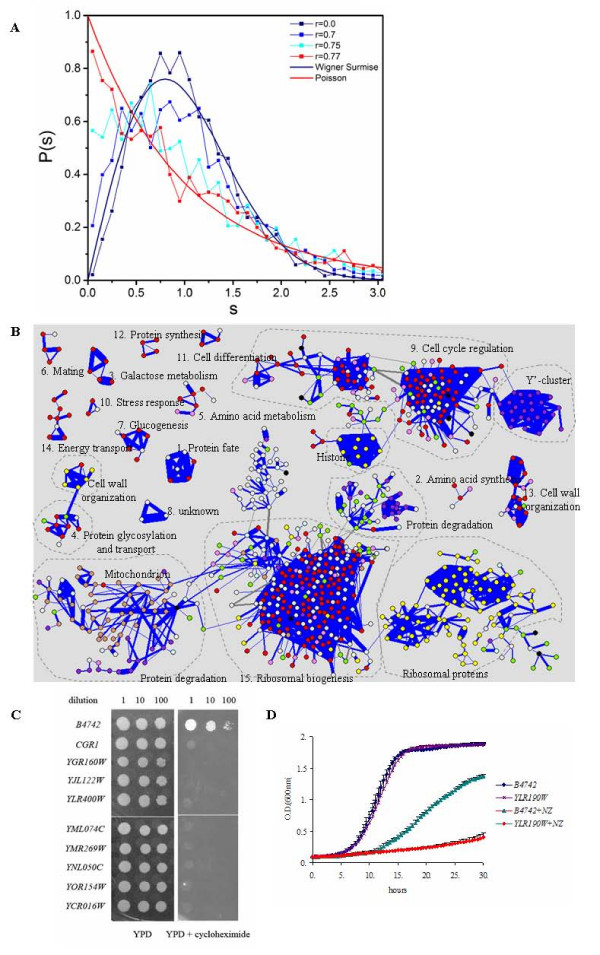
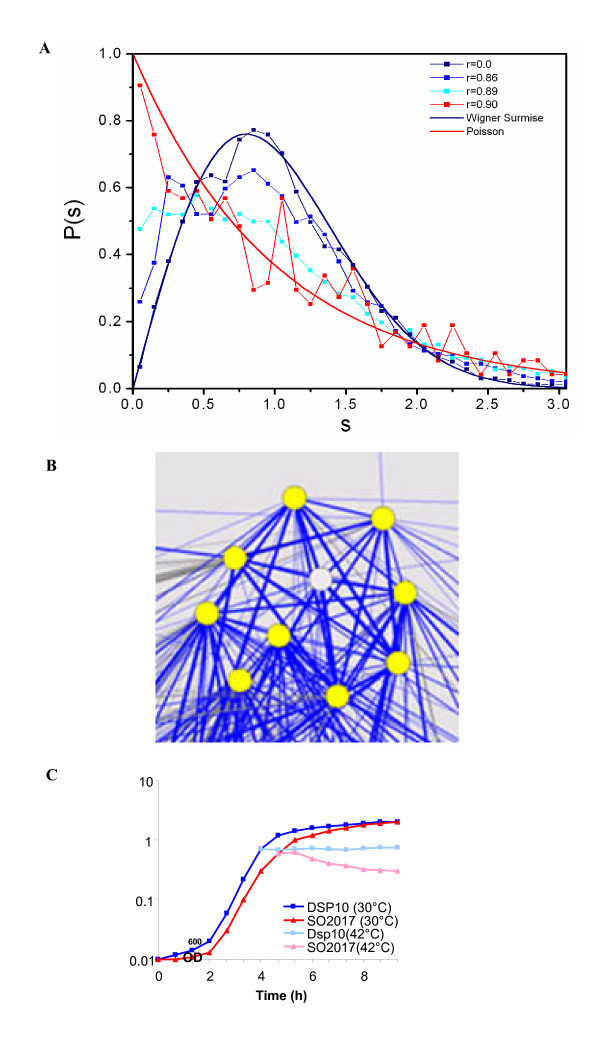
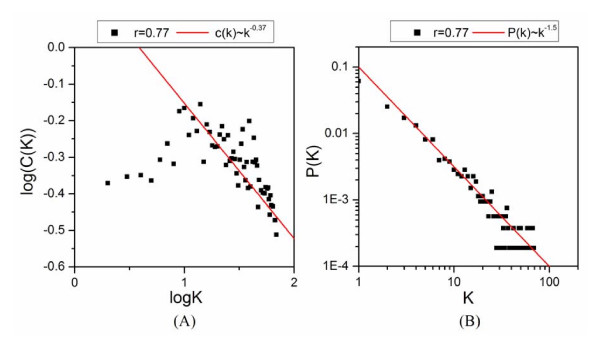
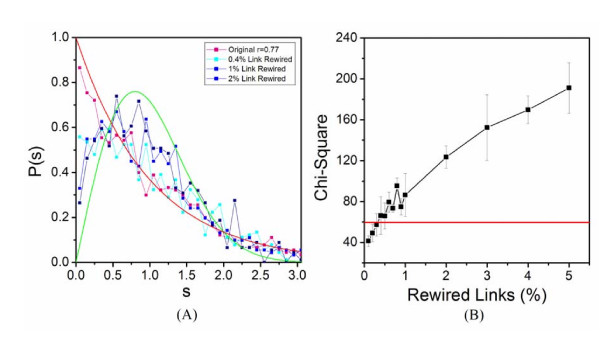
Application of random matrix theory to microarray data of S. oneidensis, E. coli, yeast, A. thaliana, Drosophila, mouse and human indicates that there is a sharp transition of nearest neighbour spacing distribution (NNSD) of correlation matrix after gradually removing certain elements insider the matrix. Testing on an in silico modular model has demonstrated that this transition can be used to determine the correlation threshold for revealing modular co-expression networks. The co-expression network derived from yeast cell cycling microarray data is supported by gene annotation. The topological properties of the resulting co-expression network agree well with the general properties of biological networks. Computational evaluations have showed that RMT approach is sensitive and robust. Furthermore, evaluation on sampled expression data of an in silico modular gene system has showed that under-sampled expressions do not affect the recovery of gene co-expression network. Moreover, the cellular roles of 215 functionally unknown genes from yeast, E. coli and S. oneidensis are predicted by the gene co-expression networks using guilt-by-association principle, many of which are supported by existing information or our experimental verification, further demonstrating the reliability of this approach for gene function prediction.
Our rigorous analysis of gene expression microarray profiles using RMT has showed that the transition of NNSD of correlation matrix of microarray profile provides a profound theoretical criterion to determine the correlation threshold for identifying gene co-expression networks.
全基因组的大规模测序开启了生物学的新时代。下一个重大挑战之一是剖析由许多单个功能模块组成的细胞网络。基于全基因组微阵列数据明确界定共表达网络很困难,而且当前方法不够稳健,与不同数据集不一致。对于了解甚少的生物体而言,这一问题尤为突出,因为可用于确定区分真实相关性与随机噪声阈值的现有生物学知识不多。随机矩阵理论(RMT)在物理学中已得到广泛且成功的应用,是一种从随机噪声中区分复杂系统中嵌入的系统特定非随机特性的强大方法。在此,我们假设RMT的通用预测也适用于生物系统,并且可以通过使用随机矩阵理论表征微阵列谱的相关矩阵来确定相关阈值。
将随机矩阵理论应用于嗜水栖热袍菌、大肠杆菌、酵母、拟南芥、果蝇、小鼠和人类的微阵列数据表明,在逐渐去除矩阵内部的某些元素后,相关矩阵的最近邻间距分布(NNSD)会发生急剧转变。在计算机模拟模块模型上的测试表明,这种转变可用于确定揭示模块共表达网络的相关阈值。从酵母细胞周期微阵列数据得出的共表达网络得到了基因注释的支持。所得共表达网络的拓扑特性与生物网络的一般特性高度吻合。计算评估表明,RMT方法灵敏且稳健。此外,对计算机模拟模块基因系统的采样表达数据的评估表明,采样不足的表达不会影响基因共表达网络的恢复。此外,利用基因共表达网络通过关联有罪原则预测了来自酵母、大肠杆菌和嗜水栖热袍菌的215个功能未知基因的细胞作用,其中许多得到了现有信息或我们实验验证的支持,进一步证明了该方法用于基因功能预测的可靠性。
我们使用RMT对基因表达微阵列谱进行的严格分析表明,微阵列谱相关矩阵的NNSD转变为确定识别基因共表达网络的相关阈值提供了一个深刻的理论标准。