McGuffin Liam J
The School of Biological Sciences, University of Reading, Whiteknights, Reading RG6 6AS, UK.
BMC Bioinformatics. 2007 Sep 18;8:345. doi: 10.1186/1471-2105-8-345.
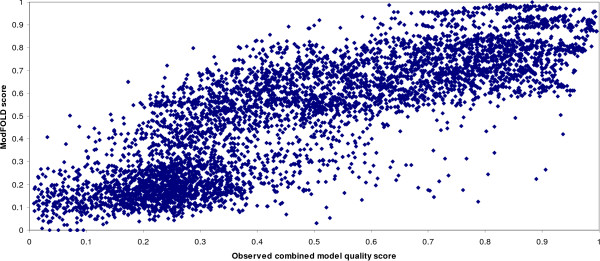
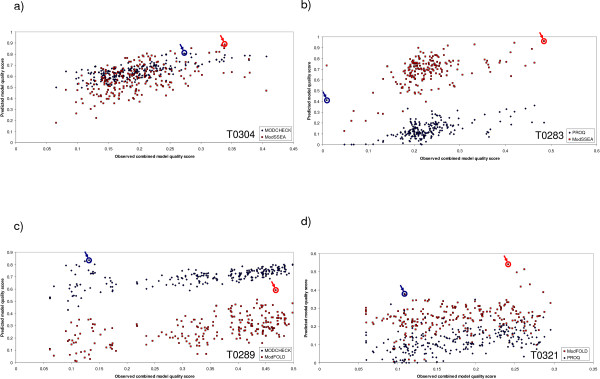
Selecting the highest quality 3D model of a protein structure from a number of alternatives remains an important challenge in the field of structural bioinformatics. Many Model Quality Assessment Programs (MQAPs) have been developed which adopt various strategies in order to tackle this problem, ranging from the so called "true" MQAPs capable of producing a single energy score based on a single model, to methods which rely on structural comparisons of multiple models or additional information from meta-servers. However, it is clear that no current method can separate the highest accuracy models from the lowest consistently. In this paper, a number of the top performing MQAP methods are benchmarked in the context of the potential value that they add to protein fold recognition. Two novel methods are also described: ModSSEA, which based on the alignment of predicted secondary structure elements and ModFOLD which combines several true MQAP methods using an artificial neural network.
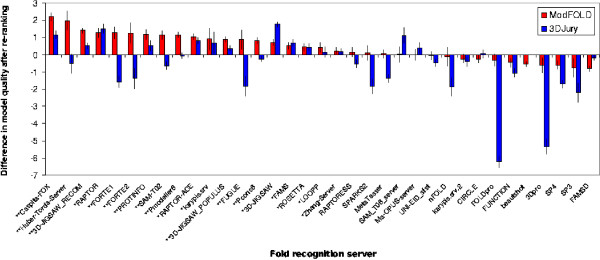
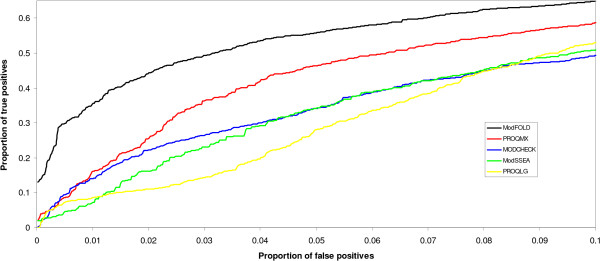
The ModSSEA method is found to be an effective model quality assessment program for ranking multiple models from many servers, however further accuracy can be gained by using the consensus approach of ModFOLD. The ModFOLD method is shown to significantly outperform the true MQAPs tested and is competitive with methods which make use of clustering or additional information from multiple servers. Several of the true MQAPs are also shown to add value to most individual fold recognition servers by improving model selection, when applied as a post filter in order to re-rank models.
MQAPs should be benchmarked appropriately for the practical context in which they are intended to be used. Clustering based methods are the top performing MQAPs where many models are available from many servers; however, they often do not add value to individual fold recognition servers when limited models are available. Conversely, the true MQAP methods tested can often be used as effective post filters for re-ranking few models from individual fold recognition servers and further improvements can be achieved using a consensus of these methods.
从多个备选模型中选择蛋白质结构的最高质量3D模型,仍然是结构生物信息学领域的一项重大挑战。已经开发了许多模型质量评估程序(MQAP),它们采用各种策略来解决这个问题,从能够基于单个模型产生单一能量分数的所谓“真正的”MQAP,到依赖于多个模型的结构比较或来自元服务器的附加信息的方法。然而,很明显,目前没有一种方法能够始终如一地将最高准确性的模型与最低准确性的模型区分开来。在本文中,一些表现最佳的MQAP方法在它们为蛋白质折叠识别增加的潜在价值的背景下进行了基准测试。还描述了两种新方法:基于预测二级结构元件比对的ModSSEA和使用人工神经网络结合多种真正MQAP方法的ModFOLD。
发现ModSSEA方法是一种有效的模型质量评估程序,可用于对来自多个服务器的多个模型进行排名,但是通过使用ModFOLD的共识方法可以获得更高的准确性。结果表明,ModFOLD方法明显优于所测试的真正MQAP,并且与使用聚类或来自多个服务器的附加信息的方法具有竞争力。当作为后过滤器应用以重新对模型进行排名时,一些真正的MQAP还通过改进模型选择为大多数单个折叠识别服务器增加了价值。
MQAP应针对其预期使用的实际背景进行适当的基准测试。基于聚类的方法是表现最佳的MQAP,当有来自多个服务器的许多模型时;然而,当可用模型有限时,它们通常不会为单个折叠识别服务器增加价值。相反,所测试的真正MQAP方法通常可以用作有效的后过滤器,用于对来自单个折叠识别服务器的少数模型进行重新排名,并且通过这些方法的共识可以实现进一步的改进。