Silverman B David
IBM Thomas J, Watson Research Center P, O, Box 218, Yorktown Heights, NY 10598, USA.
BMC Struct Biol. 2007 Nov 9;7:77. doi: 10.1186/1472-6807-7-77.
Comparison of different protein x-ray structures has previously been made in a number of different ways; for example, by visual examination, by differences in the locations of secondary structures, by explicit superposition of structural elements, e.g. alpha-carbon atom locations, or by procedures that utilize a common symmetry element or geometrical feature of the structures to be compared.
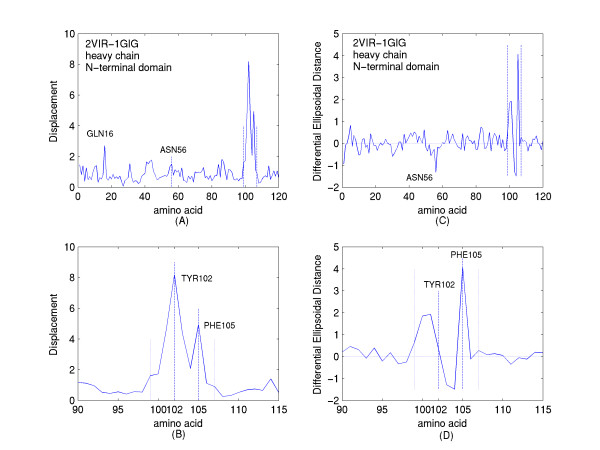
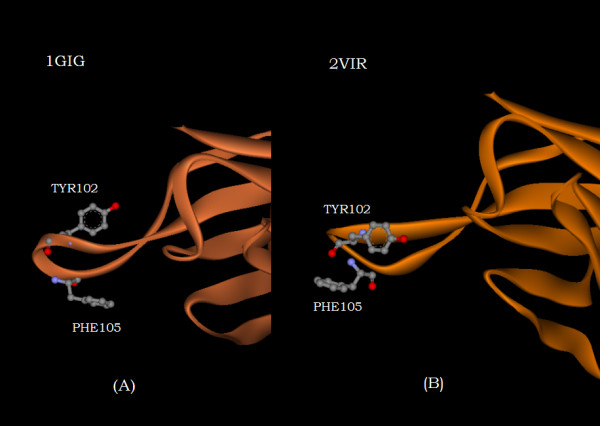
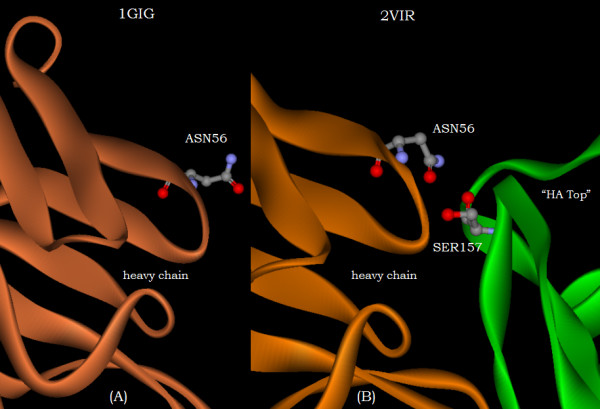
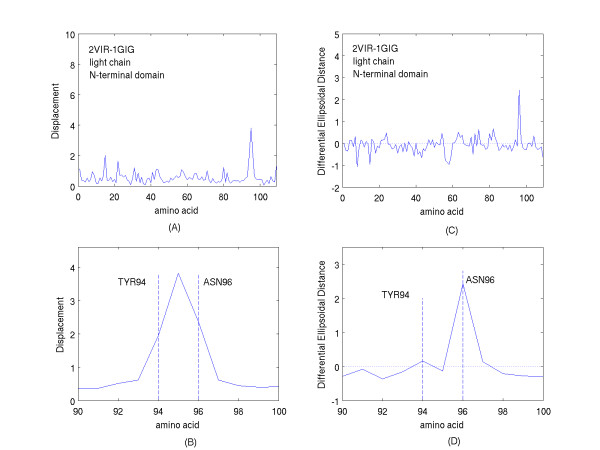
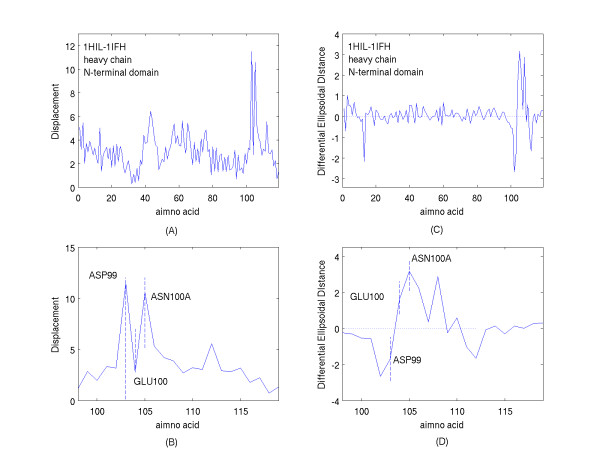
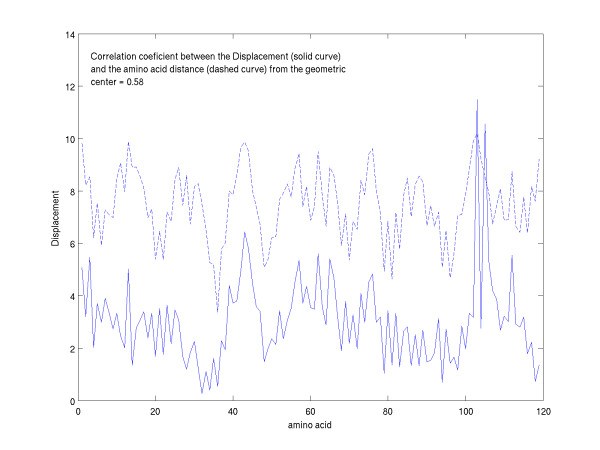
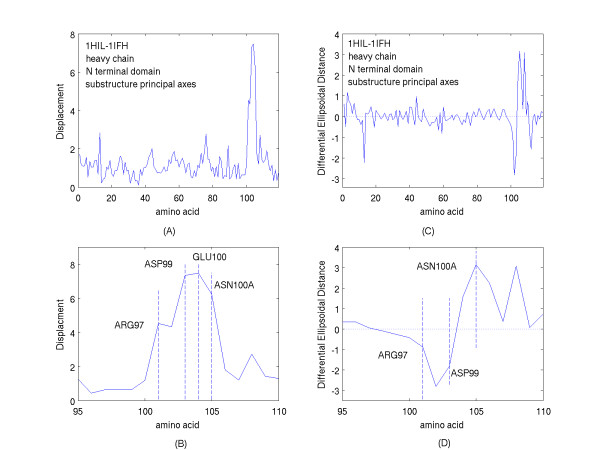
A new approach is applied to determine the structural changes that an antibody protein domain experiences upon its interaction with an antigenic target. These changes are determined with the use of two different, however comparable, sets of principal axes that are obtained by diagonalizing the second-order tensors that yield the moments-of-geometry as well as an ellipsoidal characterization of domain shape, prior to and after interaction. Determination of these sets of axes for structural comparison requires no internal symmetry features of the domains, depending solely upon their representation in three-dimensional space. This representation may involve atomic, Calpha, or residue centroid coordinates. The present analysis utilizes residue centroids. When the structural changes are minimal, the principal axes of the domains, prior to and after interaction, are essentially comparable and consequently may be used for structural comparison. When the differences of the axes cannot be neglected, but are nevertheless slight, a smaller relatively invariant substructure of the domains may be utilized for comparison. The procedure yields two distance metrics for structural comparison. First, the displacements of the residue centroids due to antigenic binding, referenced to the ellipsoidal principal axes, are noted. Second, changes in the ellipsoidal distances with respect to the non-interacting structure provide a direct measure of the spatial displacements of the residue centroids, towards either the interior or exterior of the domain.
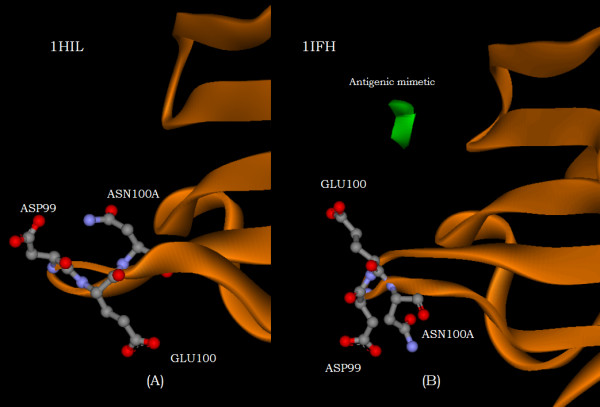
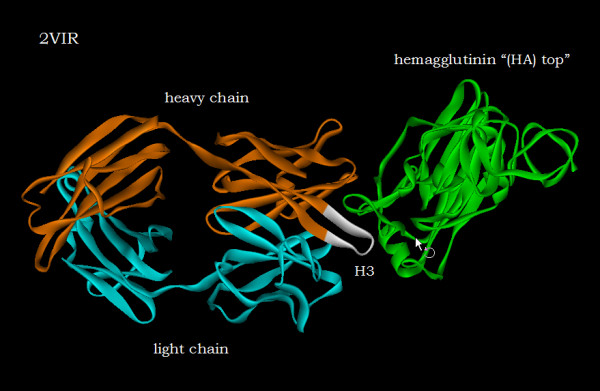
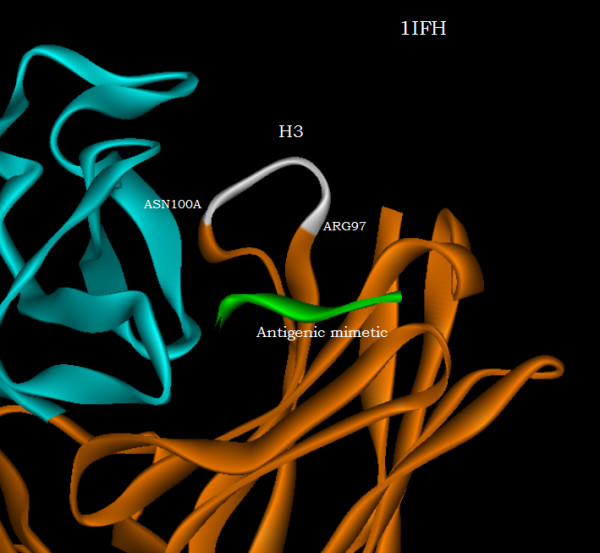
With use of x-ray data from the protein data bank (PDB), these two metrics are shown to highlight, in a manner different from before, the structural changes that are induced in the overall domains as well as in the H3 loops of the complementarity-determining regions (CDR) upon FAB antibody binding to a truncated and to a synthetic hemagglutinin viral antigenic target.
先前已通过多种不同方式对不同的蛋白质X射线结构进行比较;例如,通过目视检查、通过二级结构位置的差异、通过结构元件(如α碳原子位置)的显式叠加,或通过利用待比较结构的共同对称元素或几何特征的程序。
应用一种新方法来确定抗体蛋白结构域与抗原靶标相互作用时所经历的结构变化。这些变化通过使用两组不同但可比的主轴来确定,这两组主轴是通过对产生几何矩的二阶张量进行对角化以及在相互作用前后对结构域形状进行椭球表征而获得的。确定用于结构比较的这些轴集不需要结构域的内部对称特征,仅取决于它们在三维空间中的表示。这种表示可能涉及原子坐标、α碳原子坐标或残基质心坐标。本分析使用残基质心。当结构变化很小时,相互作用前后结构域的主轴基本可比,因此可用于结构比较。当轴的差异不可忽略但仍然很小时,可以使用结构域中较小的相对不变子结构进行比较。该程序产生用于结构比较的两个距离度量。首先,记录由于抗原结合导致的残基质心相对于椭球主轴的位移。其次,相对于非相互作用结构的椭球距离变化提供了残基质心向结构域内部或外部空间位移的直接度量。
利用来自蛋白质数据库(PDB)的X射线数据,这两个度量以与以前不同的方式突出显示了在FAB抗体与截短的和合成的血凝素病毒抗原靶标结合时,在整个结构域以及互补决定区(CDR)的H3环中诱导的结构变化。