Kaplan Shai, Itzkovitz Shalev, Shapiro Ehud
Department of Biological Chemistry, Weizmann Institute of Science, Rehovot, Israel.
PLoS Comput Biol. 2007 Nov;3(11):e235. doi: 10.1371/journal.pcbi.0030235. Epub 2007 Oct 16.
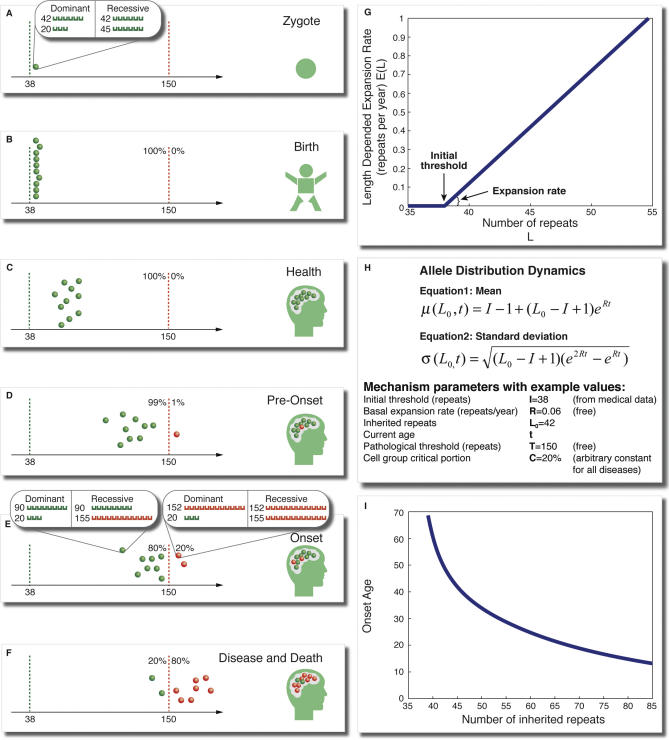
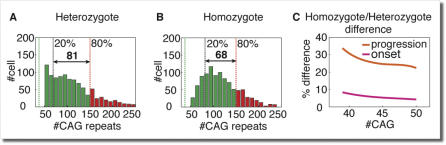
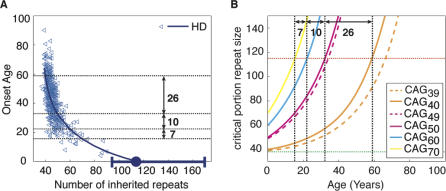
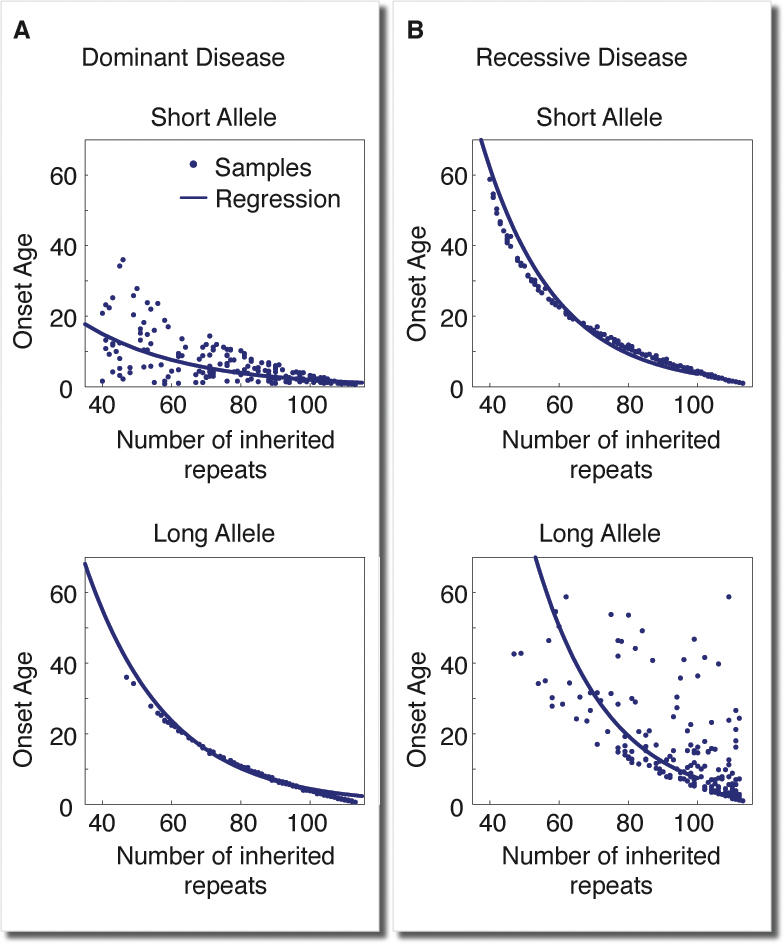
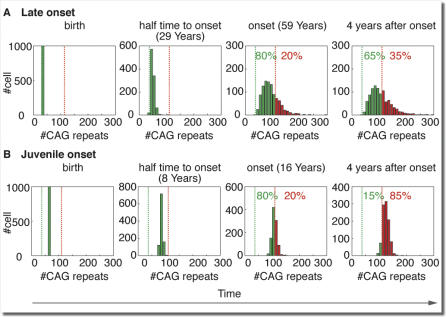
Trinucleotide hereditary diseases such as Huntington disease and Friedreich ataxia are cureless diseases associated with inheriting an abnormally large number of DNA trinucleotide repeats in a gene. The genes associated with different diseases are unrelated and harbor a trinucleotide repeat in different functional regions; therefore, it is striking that many of these diseases have similar correlations between their genotype, namely the number of inherited repeats and age of onset and progression phenotype. These correlations remain unexplained despite more than a decade of research. Although mechanisms have been proposed for several trinucleotide diseases, none of the proposals, being disease-specific, can account for the commonalities among these diseases. Here, we propose a universal mechanism in which length-dependent somatic repeat expansion occurs during the patient's lifetime toward a pathological threshold. Our mechanism uniformly explains for the first time to our knowledge the genotype-phenotype correlations common to trinucleotide disease and is well-supported by both experimental and clinical data. In addition, mathematical analysis of the mechanism provides simple explanations to a wide range of phenomena such as the exponential decrease of the age-of-onset curve, similar onset but faster progression in patients with Huntington disease with homozygous versus heterozygous mutation, and correlation of age of onset with length of the short allele but not with the long allele in Friedreich ataxia. If our proposed universal mechanism proves to be the core component of the actual mechanisms of specific trinucleotide diseases, it would open the search for a uniform treatment for all these diseases, possibly by delaying the somatic expansion process.
三核苷酸遗传性疾病,如亨廷顿舞蹈症和弗里德赖希共济失调症,是与基因中异常大量的DNA三核苷酸重复序列遗传相关的无法治愈的疾病。与不同疾病相关的基因彼此无关,且在不同的功能区域含有三核苷酸重复序列;因此,令人惊讶的是,这些疾病中的许多在其基因型(即遗传重复序列的数量)与发病年龄和疾病进展表型之间具有相似的相关性。尽管经过了十多年的研究,这些相关性仍无法得到解释。虽然已经针对几种三核苷酸疾病提出了一些机制,但这些特定于某种疾病的机制都无法解释这些疾病之间的共性。在此,我们提出一种通用机制,即在患者的一生中会发生长度依赖性的体细胞重复序列扩增,直至达到病理阈值。据我们所知,我们的机制首次统一解释了三核苷酸疾病共有的基因型 - 表型相关性,并且得到了实验和临床数据的有力支持。此外,对该机制的数学分析为广泛的现象提供了简单的解释,例如发病年龄曲线的指数下降、亨廷顿舞蹈症纯合突变患者与杂合突变患者发病相似但进展更快,以及弗里德赖希共济失调症中发病年龄与短等位基因长度相关而与长等位基因长度无关。如果我们提出的通用机制被证明是特定三核苷酸疾病实际机制的核心组成部分,那么可能通过延缓体细胞扩增过程,开启针对所有这些疾病的统一治疗方法的探索。