Bolton Judy L, Thatcher Gregory R J
Department of Medicinal Chemisry and Pharmacognosy (M/C 781), College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60612-7231, USA.
Chem Res Toxicol. 2008 Jan;21(1):93-101. doi: 10.1021/tx700191p. Epub 2007 Dec 4.
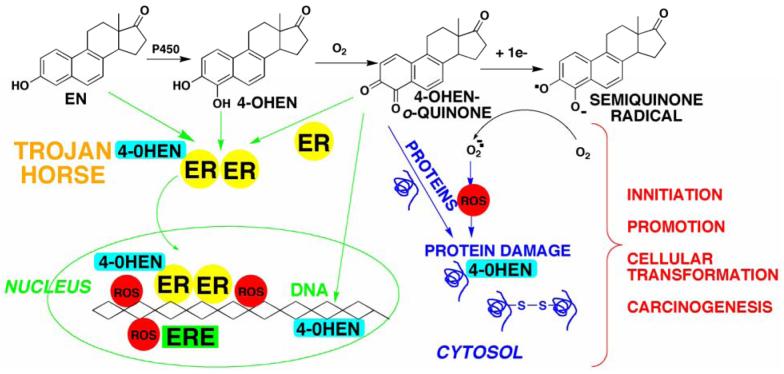
There is a clear association between the excessive exposure to estrogens and the development of cancer in hormone-sensitive tissues (breast, endometrium). It has become clear that there are likely multiple overlapping mechanisms of estrogen carcinogenesis. One major pathway is the extensively studied hormonal pathway, by which estrogen stimulates cell proliferation through nuclear estrogen receptor (ER)-mediated signaling, thus resulting in an increased risk of genomic mutations during DNA replication. A similar "nongenomic pathway", potentially involving newly discovered membrane-associated ERs, also appears to regulate extranuclear estrogen signaling pathways. This perspective is focused on a third pathway involving the metabolism of estrogens to catechols mediated by cytochrome P450 and further oxidation of these catechols to estrogen o-quinones. Oxidative enzymes, metal ions, and in some cases molecular oxygen can catalyze o-quinone formation, so that these electrophilic/redox-active quinones can cause damage within cells by alkylation and/or oxidation of cellular proteins and DNA in many tissues. It appears that the endogenous estrogen quinones primarily form unstable N3-adenine or N7-guanine DNA adducts, ultimately resulting in mutagenic apurinic sites. In contrast, equine estrogen quinones, formed from estrogens present in popular hormone replacement therapy prescriptions, generate a variety of DNA lesions, including bulky stable adducts, apurinic sites, DNA strand cleavage, and oxidation of DNA bases. DNA damage induced by these equine quinones is significantly increased in cells containing ERs, leading us to hypothesize a mechanism involving ER binding/alkylation by the catchol/quinone, resulting in a "Trojan horse". The "Trojan horse" carries the highly redox-active catechol to estrogen -sensitive genes, where high amounts of reactive oxygen species are generated, causing selective DNA damage. Our data further suggest that other key protein targets for estrogen o-quinones could be redox-sensitive enzymes (i.e, GST P1-1, QR). These proteins are involved in stress response cascades that are known to contribute to the regulation of cell proliferation and apoptosis. Finally, it has been shown that catechol estrogens can transform breast epithelial cells into a tumorigenic phenotype and that these transformed cells had differential gene expression of several genes involved in oxidative stress. Given the direct link between excessive exposure to estrogens, metabolism of estrogens, and increased risk of breast cancer, it is crucial that factors that affect the formation, reactivity, and cellular targets of estrogen quinoids be thoroughly explored.
雌激素暴露过量与激素敏感组织(乳腺、子宫内膜)癌症的发生之间存在明确关联。目前已明确,雌激素致癌可能存在多种重叠机制。一条主要途径是经过广泛研究的激素途径,雌激素通过核雌激素受体(ER)介导的信号传导刺激细胞增殖,从而在DNA复制过程中增加基因组突变风险。一条类似的“非基因组途径”,可能涉及新发现的膜相关ER,似乎也能调节核外雌激素信号通路。本文观点聚焦于第三条途径,即细胞色素P450介导雌激素代谢为儿茶酚,并将这些儿茶酚进一步氧化为雌激素邻醌。氧化酶、金属离子以及在某些情况下分子氧可催化邻醌形成,因此这些亲电/具有氧化还原活性的醌可通过烷基化和/或氧化许多组织中的细胞蛋白质和DNA在细胞内造成损伤。内源性雌激素醌似乎主要形成不稳定的N3 -腺嘌呤或N7 -鸟嘌呤DNA加合物,最终导致诱变的脱嘌呤位点。相比之下,流行的激素替代疗法处方中存在的雌激素形成的马雌激素醌会产生多种DNA损伤,包括大量稳定加合物、脱嘌呤位点、DNA链断裂以及DNA碱基氧化。这些马醌诱导的DNA损伤在含有ER的细胞中显著增加,这使我们推测存在一种机制,即儿茶酚/醌与ER结合/烷基化,从而形成一种“特洛伊木马”。“特洛伊木马”将具有高度氧化还原活性的儿茶酚携带至雌激素敏感基因,在那里产生大量活性氧,导致选择性DNA损伤。我们的数据进一步表明,雌激素邻醌的其他关键蛋白质靶点可能是对氧化还原敏感的酶(即GST P1 - 1、QR)。这些蛋白质参与应激反应级联,已知其有助于调节细胞增殖和凋亡。最后,研究表明儿茶酚雌激素可将乳腺上皮细胞转化为致瘤表型,且这些转化细胞中参与氧化应激的多个基因存在差异表达。鉴于雌激素暴露过量、雌激素代谢与乳腺癌风险增加之间的直接联系,深入探究影响雌激素醌类的形成、反应性和细胞靶点的因素至关重要。