Mackie Alexander R, Brueggemann Lioubov I, Henderson Kyle K, Shiels Aaron J, Cribbs Leanne L, Scrogin Karie E, Byron Kenneth L
Loyola University Medical Center, 2160 S. First Ave., Maywood, IL 60153, USA.
J Pharmacol Exp Ther. 2008 May;325(2):475-83. doi: 10.1124/jpet.107.135764. Epub 2008 Feb 13.
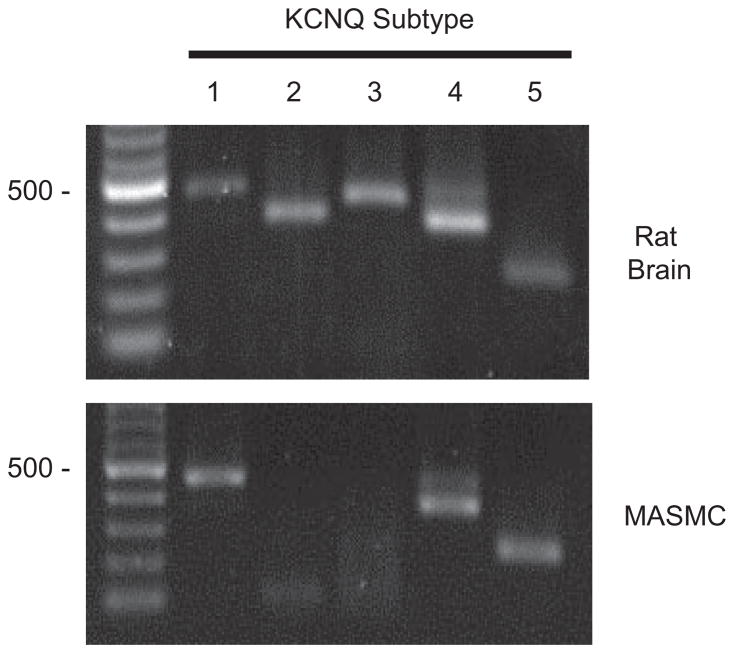
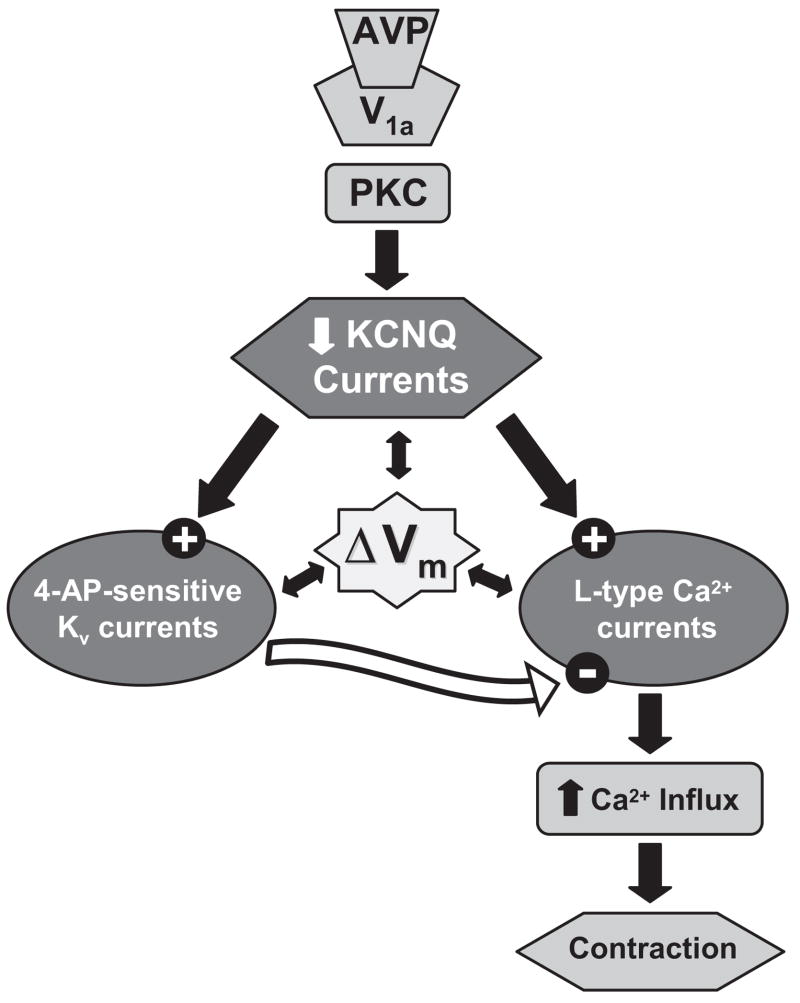
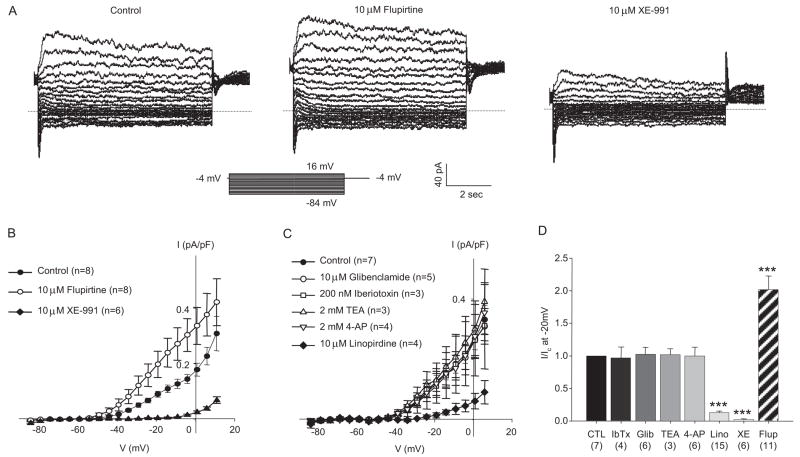
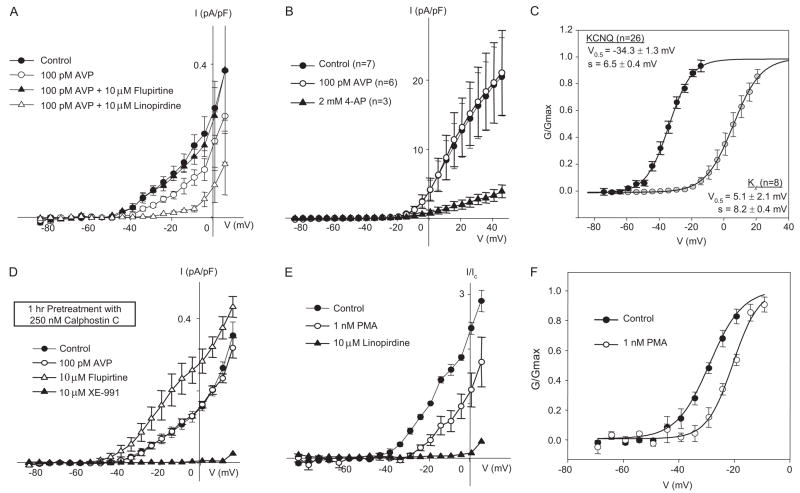
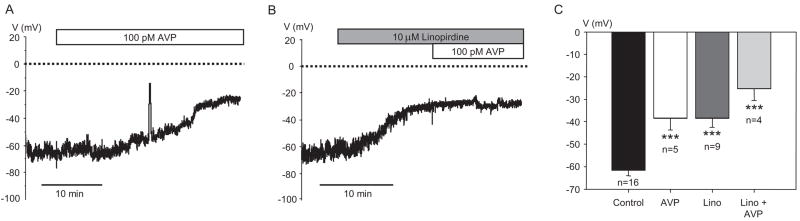
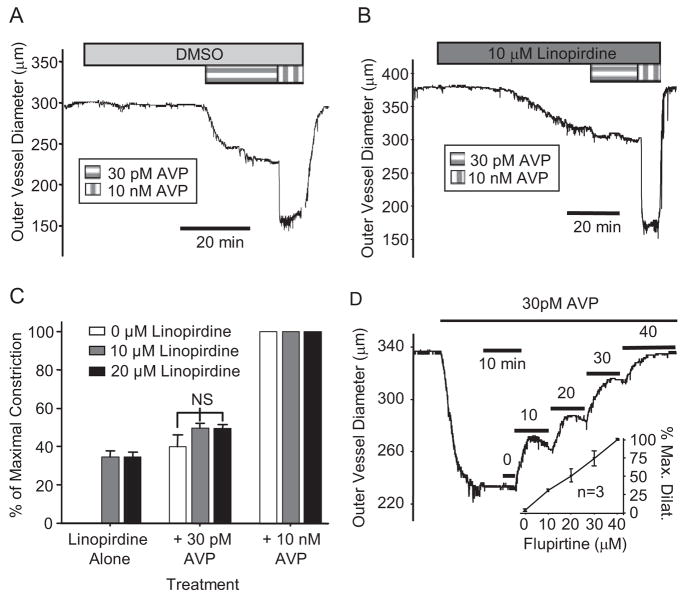
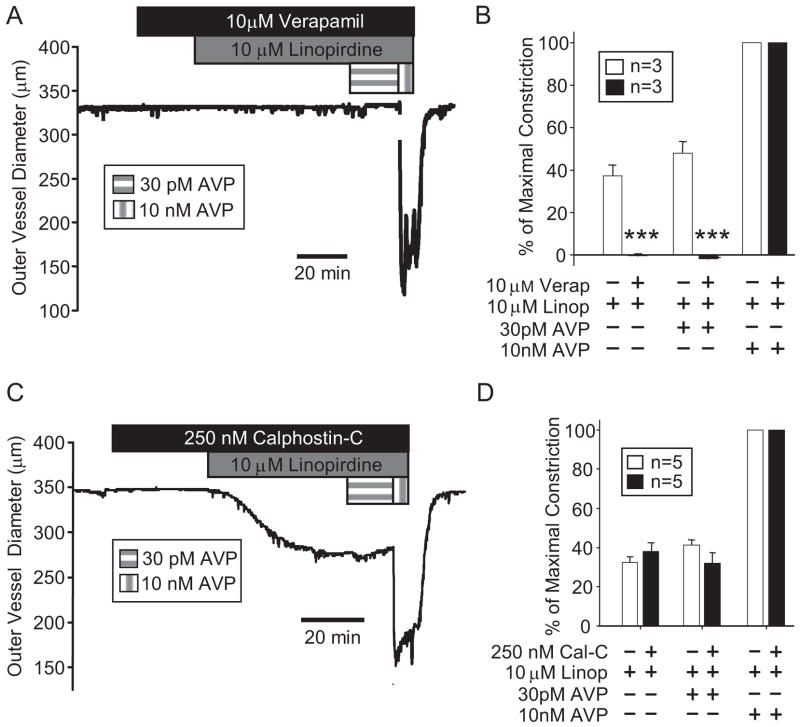
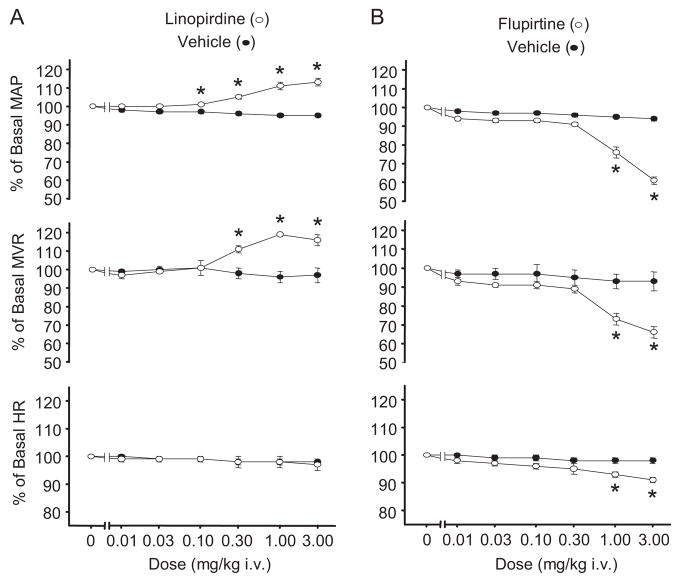
Pressor effects of the vasoconstrictor hormone arginine vasopressin (AVP), observed when systemic AVP concentrations are less than 100 pM, are important for the physiological maintenance of blood pressure, and they are also the basis for therapeutic use of vasopressin to restore blood pressure in hypotensive patients. However, the mechanisms by which circulating AVP induces arterial constriction are unclear. We examined the novel hypothesis that KCNQ potassium channels mediate the physiological vasoconstrictor actions of AVP. Reverse transcriptase polymerase chain reaction revealed expression of KCNQ1, KCNQ4, and KCNQ5 in rat mesenteric artery smooth muscle cells (MASMCs). Whole-cell perforated patch recordings of voltage-sensitive K+ (Kv) currents in freshly isolated MASMCs revealed 1,3-dihydro-1-phenyl-3,3-bis(4-pyridinylmethyl)-2H-indol-2-one (linopirdine)- and 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE-991)-sensitive KCNQ currents that were electrophysiologically and pharmacologically distinct from other Kv currents. Suppression of KCNQ currents by AVP (100 pM) was associated with significant membrane depolarization, and it was abolished by the protein kinase C (PKC) inhibitor calphostin C (250 nM). The KCNQ channel blocker linopirdine (10 microM) inhibited KCNQ currents in MASMCs, and it induced constriction of isolated rat mesenteric arteries. The vasoconstrictor responses were not additive when combined with 30 pM AVP, and they were prevented by the L-type Ca2+ channel blocker verapamil. Ethyl-N-[2-amino-6-(4-fluorophenylmethylamino)pyridin-3-yl] carbamic acid (flupirtine) significantly enhanced KCNQ currents, and it reversed constrictor responses to 30 pM AVP. In vivo, i.v. administration of linopirdine induced a dose-dependent increase in mesenteric artery resistance and blood pressure, whereas flupirtine had the opposite effects. We conclude that physiological concentrations of AVP induce mesenteric artery constriction via PKC-dependent suppression of KCNQ currents and L-type Ca2+ channel activation in MASMCs.
当全身精氨酸加压素(AVP)浓度低于100 pM时观察到的血管收缩激素AVP的升压作用,对血压的生理维持很重要,也是使用加压素恢复低血压患者血压的治疗基础。然而,循环中的AVP诱导动脉收缩的机制尚不清楚。我们研究了一个新的假说,即KCNQ钾通道介导AVP的生理性血管收缩作用。逆转录聚合酶链反应显示大鼠肠系膜动脉平滑肌细胞(MASMCs)中KCNQ1、KCNQ4和KCNQ5的表达。对新鲜分离的MASMCs中电压敏感性钾离子(Kv)电流进行的全细胞膜片穿孔记录显示,1,3 - 二氢 - 1 - 苯基 - 3,3 - 双(4 - 吡啶基甲基)- 2H - 吲哚 - 2 - 酮(利诺吡啶)和10,10 - 双(4 - 吡啶基甲基)- 9(10H)- 蒽酮(XE - 991)敏感的KCNQ电流在电生理学和药理学上与其他Kv电流不同。AVP(100 pM)对KCNQ电流的抑制与显著的膜去极化相关,并且被蛋白激酶C(PKC)抑制剂钙泊三醇(250 nM)消除。KCNQ通道阻滞剂利诺吡啶(10 μM)抑制MASMCs中的KCNQ电流,并诱导离体大鼠肠系膜动脉收缩。当与30 pM AVP联合使用时,血管收缩反应没有叠加,并且被L型钙通道阻滞剂维拉帕米阻止。乙基 - N - [2 - 氨基 - 6 - (4 - 氟苯基甲基氨基)吡啶 - 3 - 基]氨基甲酸酯(氟吡汀)显著增强KCNQ电流,并逆转对30 pM AVP的收缩反应。在体内,静脉注射利诺吡啶导致肠系膜动脉阻力和血压呈剂量依赖性增加,而氟吡汀则有相反的作用。我们得出结论,生理浓度的AVP通过PKC依赖的对MASMCs中KCNQ电流的抑制和L型钙通道激活诱导肠系膜动脉收缩。