Turleau Catherine
Cytogénétique AP-HP et Inserm U781, Université Paris Descartes, Hôpital Necker-Enfants Malades, 75015 Paris, France.
Orphanet J Rare Dis. 2008 Feb 19;3:4. doi: 10.1186/1750-1172-3-4.
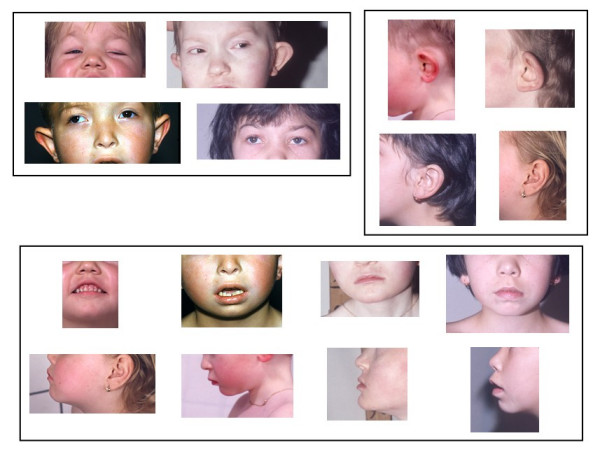
Monosomy 18p refers to a chromosomal disorder resulting from the deletion of all or part of the short arm of chromosome 18. The incidence is estimated to be about 1:50,000 live-born infants. In the commonest form of the disorder, the dysmorphic syndrome is very moderate and non-specific. The main clinical features are short stature, round face with short philtrum, palpebral ptosis and large ears with detached pinnae. Intellectual deficiency is mild to moderate. A small subset of patients, about 10-15 percent of cases, present with severe brain/facial malformations evocative of holoprosencephaly spectrum disorders. In two-thirds of the cases, the 18p- syndrome is due to a mere terminal deletion occurring de novo, in one-third the following are possible: a de novo translocation with loss of 18p, malsegregation of a parental translocation or inversion, or a ring chr18. Parental transmission of the 18p- syndrome has been reported. Cytogenetic analysis is necessary to make a definite diagnosis. Recurrence risk for siblings is low in de novo deletions and translocations, but is significant if a parental rearrangement is present. Deletion 18p can be detected prenatally by amniocentesis or chorionic villus sampling and cytogenetic testing. Differential diagnosis may include a wide number of syndromes with short stature and mild intellectual deficiency. In young children, deletion 18p syndrome may be vaguely evocative of either Turner syndrome or trisomy 21. No specific treatment exists but speech therapy and early educational programs may help to improve the performances of the children. Except for the patients with severe brain malformations, the life expectancy does not seem significantly reduced.
18号染色体短臂单体综合征是一种由于18号染色体短臂全部或部分缺失所致的染色体疾病。其发病率估计约为1:50000活产婴儿。在该疾病最常见的形式中,畸形综合征非常轻微且不具特异性。主要临床特征为身材矮小、圆脸且人中短、眼睑下垂以及耳朵大且耳廓分离。智力缺陷为轻度至中度。一小部分患者(约10% - 15%的病例)表现出严重的脑/面部畸形,提示全前脑谱系障碍。在三分之二的病例中,18p - 综合征是由于单纯的新发末端缺失,另外三分之一可能有以下情况:新发易位伴18p缺失、亲代易位或倒位的错误分离,或18号环状染色体。已有18p - 综合征亲代传递的报道。进行明确诊断需要细胞遗传学分析。新发缺失和易位时,同胞的复发风险较低,但如果存在亲代重排则风险显著。18p缺失可通过羊膜穿刺术或绒毛取样及细胞遗传学检测在产前检测到。鉴别诊断可能包括多种身材矮小和轻度智力缺陷的综合征。在幼儿中,18p缺失综合征可能会模糊地让人联想到特纳综合征或21三体综合征。目前尚无特异性治疗方法,但言语治疗和早期教育项目可能有助于提高患儿的表现。除了患有严重脑畸形的患者外,预期寿命似乎没有明显缩短。