Oleksyk Taras K, Zhao Kai, De La Vega Francisco M, Gilbert Dennis A, O'Brien Stephen J, Smith Michael W
Laboratory of Genomic Diversity, National Cancer Institute at Frederick, Frederick, Maryland, USA.
PLoS One. 2008 Mar 5;3(3):e1712. doi: 10.1371/journal.pone.0001712.
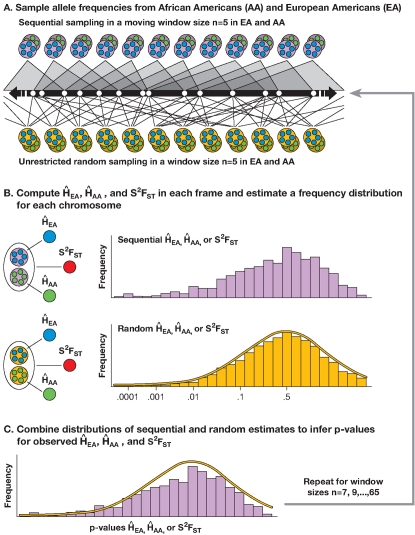
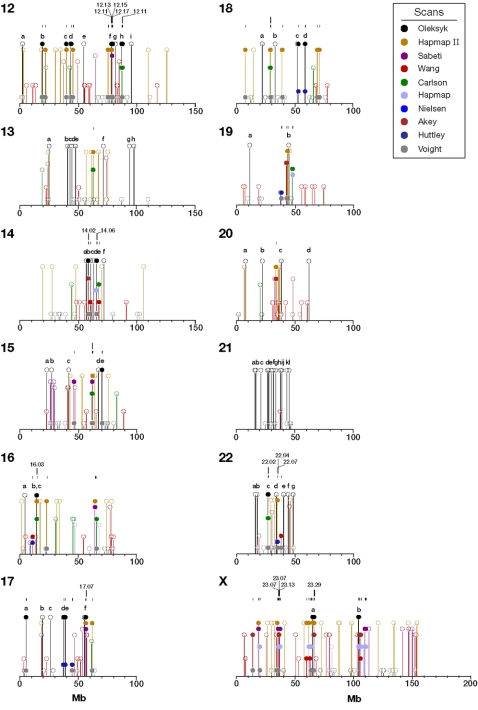
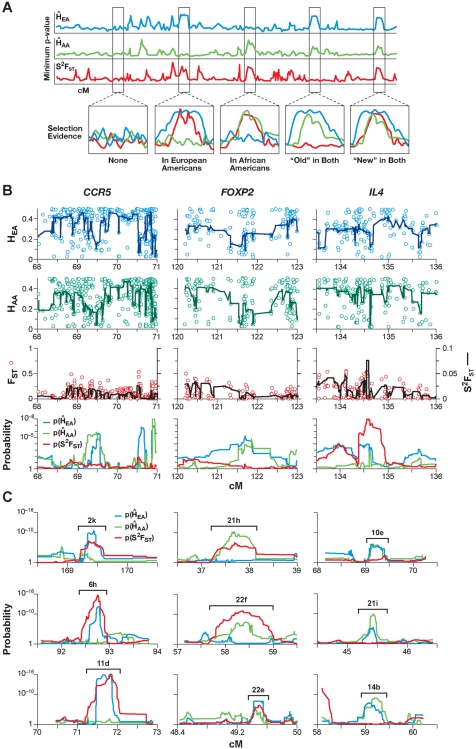
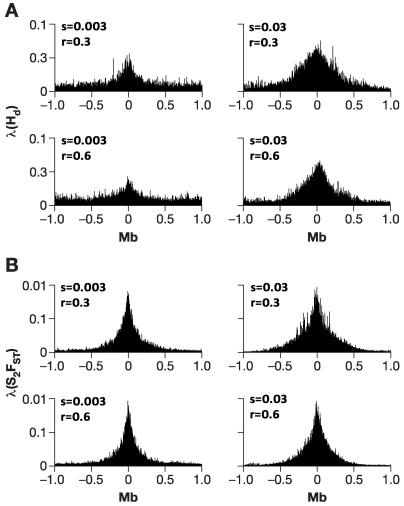
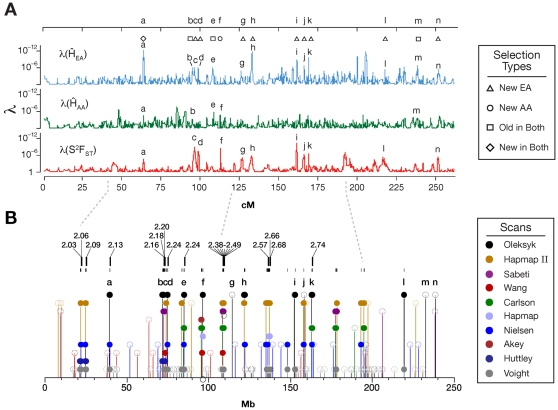
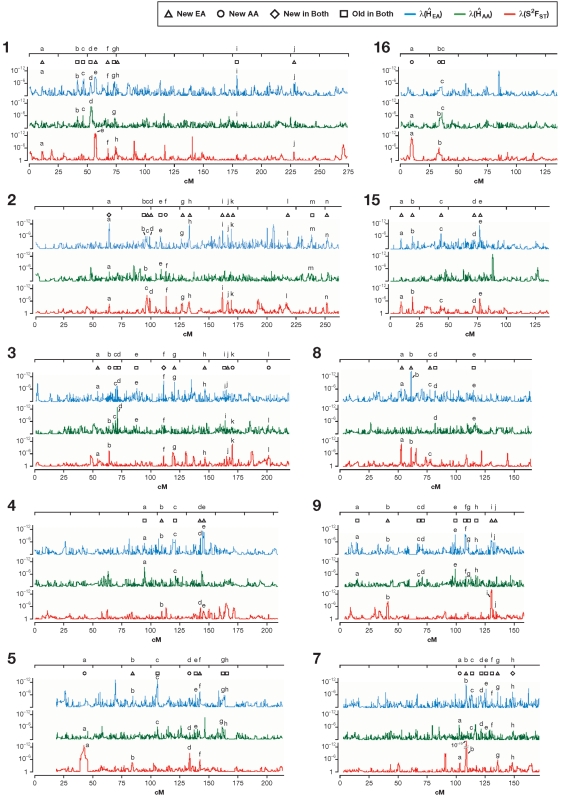
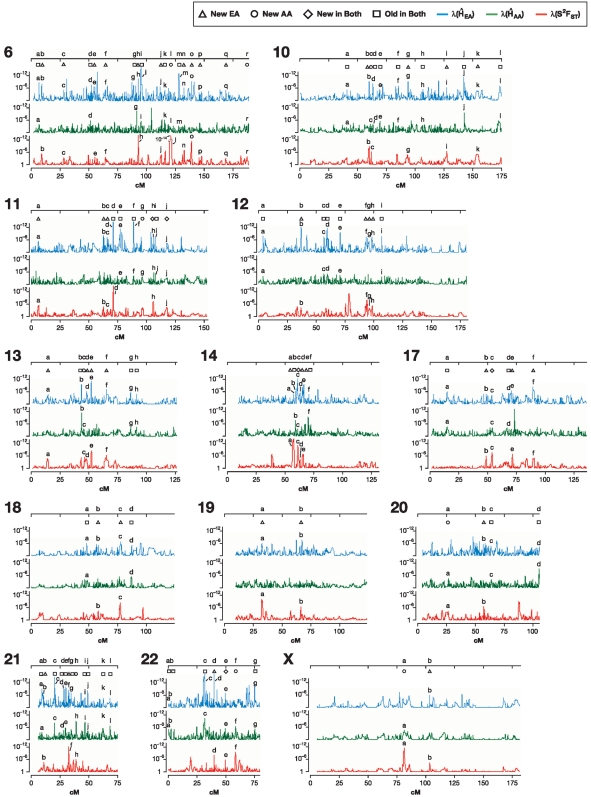
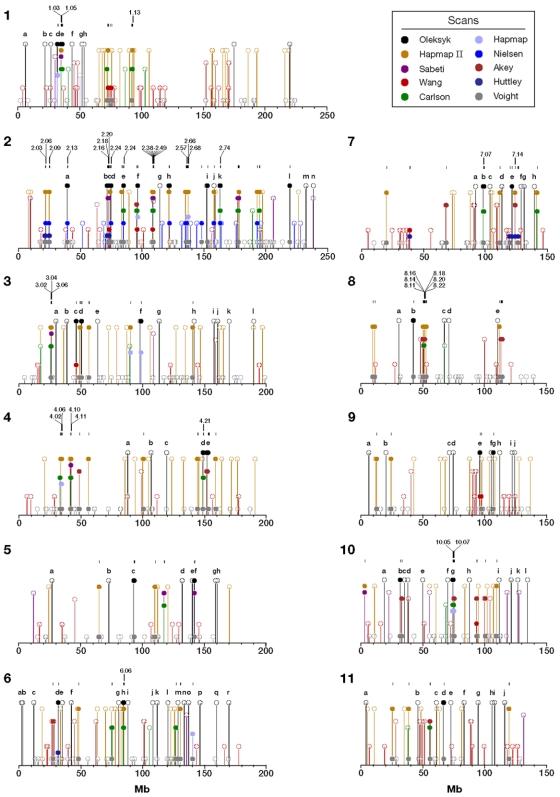
When a selective sweep occurs in the chromosomal region around a target gene in two populations that have recently separated, it produces three dramatic genomic consequences: 1) decreased multi-locus heterozygosity in the region; 2) elevated or diminished genetic divergence (F(ST)) of multiple polymorphic variants adjacent to the selected locus between the divergent populations, due to the alternative fixation of alleles; and 3) a consequent regional increase in the variance of F(ST) (S(2)F(ST)) for the same clustered variants, due to the increased alternative fixation of alleles in the loci surrounding the selection target. In the first part of our study, to search for potential targets of directional selection, we developed and validated a resampling-based computational approach; we then scanned an array of 31 different-sized moving windows of SNP variants (5-65 SNPs) across the human genome in a set of European and African American population samples with 183,997 SNP loci after correcting for the recombination rate variation. The analysis revealed 180 regions of recent selection with very strong evidence in either population or both. In the second part of our study, we compared the newly discovered putative regions to those sites previously postulated in the literature, using methods based on inspecting patterns of linkage disequilibrium, population divergence and other methodologies. The newly found regions were cross-validated with those found in nine other studies that have searched for selection signals. Our study was replicated especially well in those regions confirmed by three or more studies. These validated regions were independently verified, using a combination of different methods and different databases in other studies, and should include fewer false positives. The main strength of our analysis method compared to others is that it does not require dense genotyping and therefore can be used with data from population-based genome SNP scans from smaller studies of humans or other species.
当在两个最近分离的群体中,围绕一个目标基因的染色体区域发生选择性清除时,会产生三个显著的基因组后果:1)该区域多位点杂合性降低;2)由于等位基因的交替固定,在分化群体之间,与所选位点相邻的多个多态性变体的遗传分化(F(ST))升高或降低;3)由于选择目标周围位点上等位基因交替固定的增加,对于相同的聚类变体,F(ST)的方差(S(2)F(ST))会随之出现区域性增加。在我们研究的第一部分,为了寻找定向选择的潜在目标,我们开发并验证了一种基于重采样的计算方法;然后,在一组欧洲裔和非裔美国人的群体样本中,对183,997个单核苷酸多态性(SNP)位点进行校正以消除重组率变异后,我们扫描了跨越人类基因组的一系列31个不同大小的SNP变体移动窗口(5 - 65个SNP)。分析揭示了180个近期选择区域,在一个群体或两个群体中都有非常有力的证据。在我们研究的第二部分,我们使用基于检查连锁不平衡模式、群体分化及其他方法,将新发现的假定区域与文献中先前假定的那些位点进行了比较。新发现的区域与其他九项寻找选择信号的研究中发现的区域进行了交叉验证。我们的研究在那些被三项或更多研究证实的区域中重复验证效果特别好。这些经过验证的区域在其他研究中使用不同方法和不同数据库的组合进行了独立验证,应该包含较少的假阳性结果。与其他方法相比,我们分析方法的主要优势在于它不需要密集的基因分型,因此可用于来自人类或其他物种较小研究的基于群体的基因组SNP扫描数据。