Binenbaum Gil, McDonald-McGinn Donna M, Zackai Elaine H, Walker B Michael, Coleman Karlene, Mach Amy M, Adam Margaret, Manning Melanie, Alcorn Deborah M, Zabel Carrie, Anderson Dennis R, Forbes Brian J
Department of Pediatric Ophthalmology, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA.
Am J Med Genet A. 2008 Apr 1;146A(7):904-9. doi: 10.1002/ajmg.a.32156.
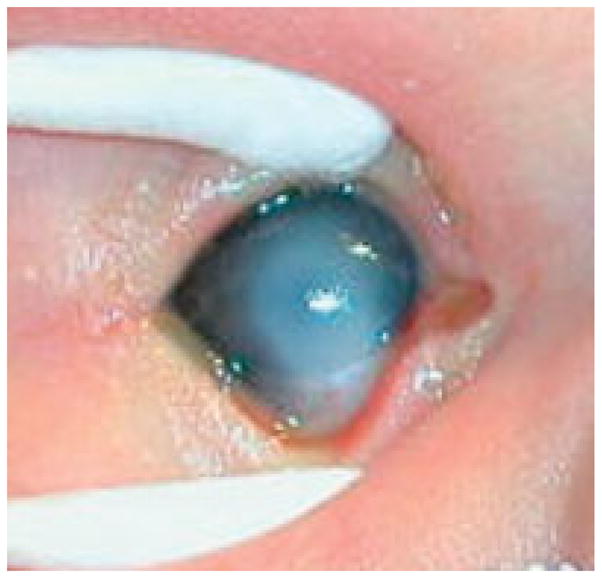
Reported ocular findings in the 22q11.2 deletion syndrome (which encompasses the phenotypes of DiGeorge, velocardiofacial, and Takao (conotruncal-anomaly-face) syndromes) have included posterior embryotoxon (prominent, anteriorly displaced Schwalbe's line at the corneal limbus or edge), retinal vascular tortuosity, eyelid hooding, strabismus, and astigmatism. We present seven 22q11.2 patients from multiple centers with sclerocornea, an eye finding previously unreported in the literature. Four boys and three girls were identified with sclerocornea, systemic DGS/VCFS findings, and fluorescence in situ hybridization (FISH)-confirmed microdeletion at chromosome 22q11.2. FISH diagnosis was perinatal in six patients but at 2 years of age in one child. Sclerocornea was bilateral in five patients. Findings included descemetocele (five eyes), microophthalmos (one eye), iridocorneal adhesions (one bilateral case), and severe anterior segment dysgenesis (one eye). Two patients underwent bilateral corneal transplantation; another two were scheduled for possible unilateral transplant. Sclerocornea is a static congenital condition in which the cornea is opaque and vascularized and resembles the sclera. The novel finding of sclerocornea suggests that a genetic locus at 22q11.2 may be involved in anterior segment embryogenesis. In most of our patients, the diagnostic process was underway, but in one patient 22q11.2 deletion was not suspected until after the child had already been undergoing treatment for sclerocornea for 2 years. Sclerocornea should be added to the clinical manifestations of the 22q11.2 deletion syndrome. Ophthalmologists diagnosing sclerocornea in children with systemic findings suggestive of 22q11.2 deletion should ensure appropriate genetic referral.
据报道,22q11.2缺失综合征(涵盖迪乔治综合征、腭心面综合征和高尾综合征(圆锥动脉干异常面容综合征)的表型)的眼部表现包括后胚胎环(角膜缘或边缘处突出的、向前移位的施瓦贝线)、视网膜血管迂曲、眼睑下垂、斜视和散光。我们展示了来自多个中心的7例22q11.2缺失综合征患者,他们患有巩膜化角膜,这是一种此前文献中未报道过的眼部表现。确定有4名男孩和3名女孩患有巩膜化角膜、系统性22q11.2缺失综合征/腭心面综合征表现以及荧光原位杂交(FISH)确认的22号染色体q11.2区域微缺失。6例患者的FISH诊断在围产期进行,但1例患儿在2岁时确诊。5例患者的巩膜化角膜为双侧性。表现包括角膜后弹力层膨出(5只眼)、小眼球(1只眼)、虹膜角膜粘连(1例双侧病例)和严重眼前段发育异常(1只眼)。2例患者接受了双侧角膜移植;另外2例计划进行单侧移植。巩膜化角膜是一种静止性先天性疾病,其角膜不透明且血管化,类似巩膜。巩膜化角膜这一新发现表明,22q11.2的一个基因位点可能参与眼前段胚胎发育。在我们的大多数患者中,诊断过程正在进行,但1例患者在接受巩膜化角膜治疗2年后才被怀疑存在22q11.2缺失。巩膜化角膜应被添加到22q11.2缺失综合征的临床表现中。眼科医生在诊断患有提示22q11.2缺失的系统性表现的儿童巩膜化角膜时,应确保进行适当的基因转诊。