Ashley Neil, O'Rourke Anthony, Smith Conrad, Adams Susan, Gowda Vasantha, Zeviani Massimo, Brown Garry K, Fratter Carl, Poulton Joanna
Nuffield Department of Obstetrics and Gynaecology, University of Oxford, The Women's Centre, John Radcliffe Hospital, Oxford OX3 9DU, UK.
Hum Mol Genet. 2008 Aug 15;17(16):2496-506. doi: 10.1093/hmg/ddn150. Epub 2008 May 16.
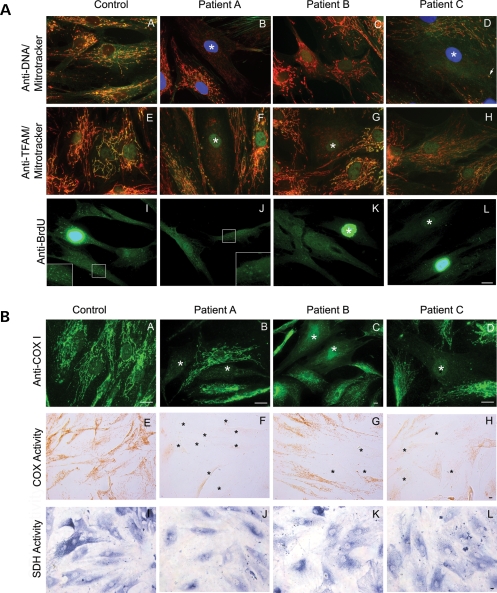
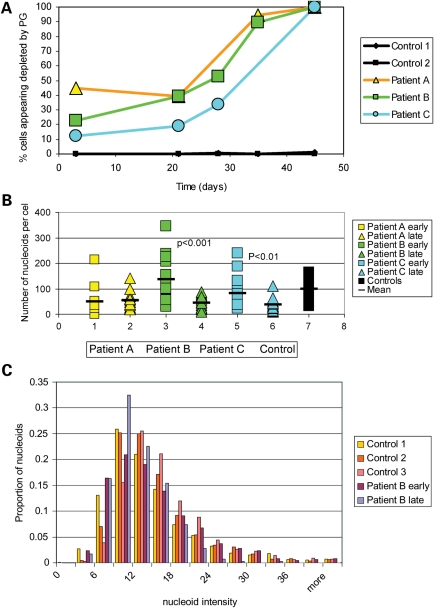
We investigated clinical and cellular phenotypes of 24 children with mutations in the catalytic (alpha) subunit of the mitochondrial DNA (mtDNA) gamma polymerase (POLG1). Twenty-one had Alpers syndrome, the commonest severe POLG1 autosomal recessive phenotype, comprising hepatoencephalopathy and often mtDNA depletion. The cellular mtDNA content reflected the genotype more closely than did clinical features. Patients with tissue depletion of mtDNA all had at least one allele with either a missense mutation in a catalytic domain or a nonsense mutation. Four out of 12 patients exhibited a progressive, mosaic pattern of mtDNA depletion in cultured fibroblasts. All these patients had mutations in a catalytic domain in both POLG1 alleles, in either the polymerase or exonuclease domain or both. The tissue mtDNA content of patients who had two linker mutations was normal, and their phenotypes the mildest. Epilepsy and/or movement disorder were major features in all 21. Previous studies have implicated replication stalling as a mechanism for mtDNA depletion. The mosaic cellular depletion that we have demonstrated in cell cultures may be a manifestation of severe replication stalling. One patient with a severe cellular and clinical phenotype was a compound heterozygote with POLG1 mutations in the polymerase and exonuclease domain intrans. This suggests that POLG1 requires both polymerase and 3'-5' exonuclease activity in the same molecule. This is consistent with current functional models for eukaryotic DNA polymerases, which alternate between polymerizing and editing modes, as determined by competition between these two active sites for the 3' end of the DNA.
我们研究了24名线粒体DNA(mtDNA)γ聚合酶(POLG1)催化(α)亚基发生突变的儿童的临床和细胞表型。其中21名患有阿尔珀斯综合征,这是最常见的严重POLG1常染色体隐性表型,包括肝性脑病且常伴有mtDNA耗竭。细胞mtDNA含量比临床特征更能紧密反映基因型。mtDNA出现组织耗竭的患者均至少有一个等位基因存在催化结构域的错义突变或无义突变。12名患者中有4名在培养的成纤维细胞中表现出mtDNA耗竭的进行性、嵌合模式。所有这些患者的两个POLG1等位基因的催化结构域均有突变,位于聚合酶结构域或核酸外切酶结构域或两者皆有。有两个连接子突变的患者的组织mtDNA含量正常,且其表型最为轻微。癫痫和/或运动障碍是所有21名患者的主要特征。先前的研究认为复制停滞是mtDNA耗竭的一种机制。我们在细胞培养中所证明的嵌合细胞耗竭可能是严重复制停滞的一种表现。一名具有严重细胞和临床表型的患者是一种复合杂合子,其POLG1在聚合酶和核酸外切酶结构域存在反式突变。这表明POLG1在同一分子中既需要聚合酶活性又需要3'-5'核酸外切酶活性。这与真核DNA聚合酶的当前功能模型一致,真核DNA聚合酶在聚合和编辑模式之间交替,这由这两个活性位点对DNA 3'末端的竞争所决定。