de Souza Gustavo A, Målen Hiwa, Søfteland Tina, Saelensminde Gisle, Prasad Swati, Jonassen Inge, Wiker Harald G
Section for Microbiology and Immunology, The Gade Institute, University of Bergen, Bergen, Norway.
BMC Genomics. 2008 Jul 2;9:316. doi: 10.1186/1471-2164-9-316.
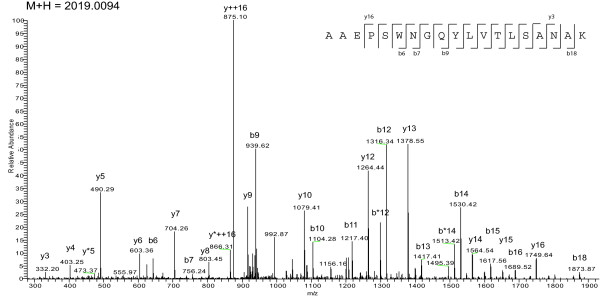
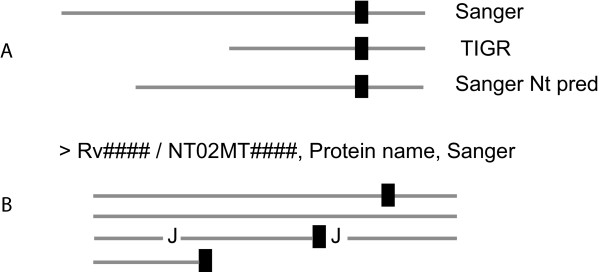
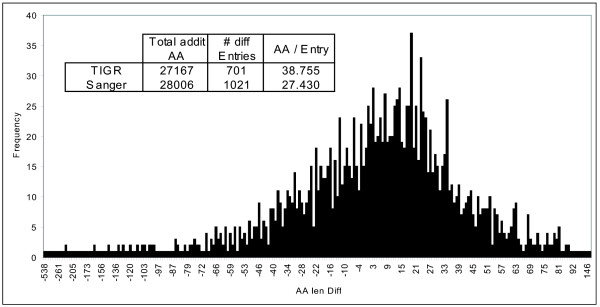
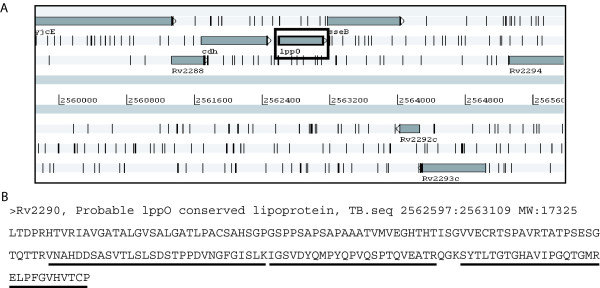
While the genomic annotations of diverse lineages of the Mycobacterium tuberculosis complex are available, divergences between gene prediction methods are still a challenge for unbiased protein dataset generation. M. tuberculosis gene annotation is an example, where the most used datasets from two independent institutions (Sanger Institute and Institute of Genomic Research-TIGR) differ up to 12% in the number of annotated open reading frames, and 46% of the genes contained in both annotations have different start codons. Such differences emphasize the importance of the identification of the sequence of protein products to validate each gene annotation including its sequence coding area.
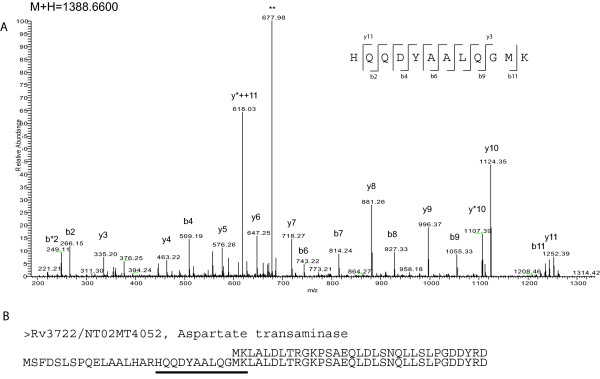
With this objective, we submitted a culture filtrate sample from M. tuberculosis to a high-accuracy LTQ-Orbitrap mass spectrometer analysis and applied refined N-terminal prediction to perform comparison of two gene annotations. From a total of 449 proteins identified from the MS data, we validated 35 tryptic peptides that were specific to one of the two datasets, representing 24 different proteins. From those, 5 proteins were only annotated in the Sanger database. In the remaining proteins, the observed differences were due to differences in annotation of transcriptional start sites.
Our results indicate that, even in a less complex sample likely to represent only 10% of the bacterial proteome, we were still able to detect major differences between different gene annotation approaches. This gives hope that high-throughput proteomics techniques can be used to improve and validate gene annotations, and in particular for verification of high-throughput, automatic gene annotations.
虽然结核分枝杆菌复合群不同谱系的基因组注释已有,但基因预测方法之间的差异仍是生成无偏差蛋白质数据集的一个挑战。结核分枝杆菌的基因注释就是一个例子,两个独立机构(桑格研究所和基因组研究所-TIGR)最常用的数据集在注释的开放阅读框数量上相差高达12%,且两个注释中包含的46%的基因具有不同的起始密码子。这些差异凸显了鉴定蛋白质产物序列以验证每个基因注释(包括其序列编码区)的重要性。
出于这一目的,我们将一份结核分枝杆菌的培养滤液样本提交给高精度LTQ-轨道阱质谱仪分析,并应用改进的N端预测来比较两种基因注释。从质谱数据鉴定出的总共449种蛋白质中,我们验证了35种胰蛋白酶肽段,它们是两个数据集中某一个所特有的,代表24种不同的蛋白质。其中,有5种蛋白质仅在桑格数据库中有注释。在其余蛋白质中,观察到的差异是由于转录起始位点注释的不同。
我们的结果表明,即使在一个可能仅代表细菌蛋白质组10%的不太复杂的样本中,我们仍能够检测到不同基因注释方法之间的主要差异。这让人们有希望利用高通量蛋白质组学技术来改进和验证基因注释,特别是用于验证高通量自动基因注释。