Yue Feng, Zhang Meng, Tang Jijun
Department of Computer Science and Engineering, University of South Carolina, Columbia, SC 29208, USA.
BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S15. doi: 10.1186/1471-2164-9-S2-S15.
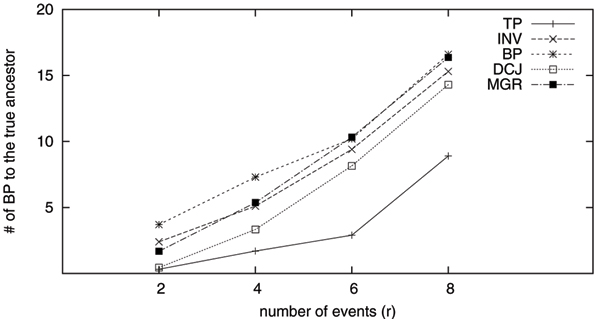
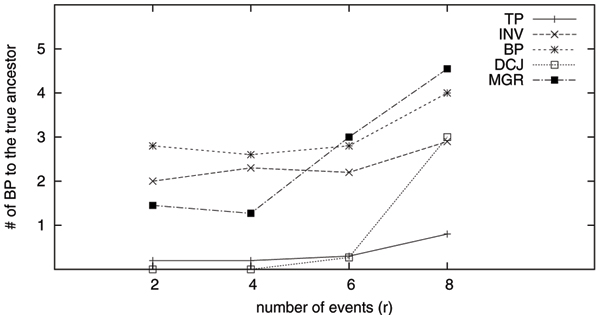
Because of the advent of high-throughput sequencing and the consequent reduction in the cost of sequencing, many organisms have been completely sequenced and most of their genes identified. It thus has become possible to represent whole genomes as ordered lists of gene identifiers and to study the rearrangement of these entities through computational means. As a result, genome rearrangement data has attracted increasing attentions from both biologists and computer scientists as a new type of data for phylogenetic analysis. The main events of genome rearrangements include inversions, transpositions and transversions. To date, GRAPPA and MGR are the most accurate methods for rearrangement phylogeny, both assuming inversion as the only event. However, due to the complexity of computing transposition distance, it is very difficult to analyze datasets when transpositions are dominant.
We extend GRAPPA to handle transpositions. The new method is named GRAPPA-TP, with two major extensions: a heuristic method to estimate transposition distance, and a new transposition median solver for three genomes. Although GRAPPA-TP uses a greedy approach to compute the transposition distance, it is very accurate when genomes are relatively close. The new GRAPPA-TP is available from http://phylo.cse.sc.edu/.
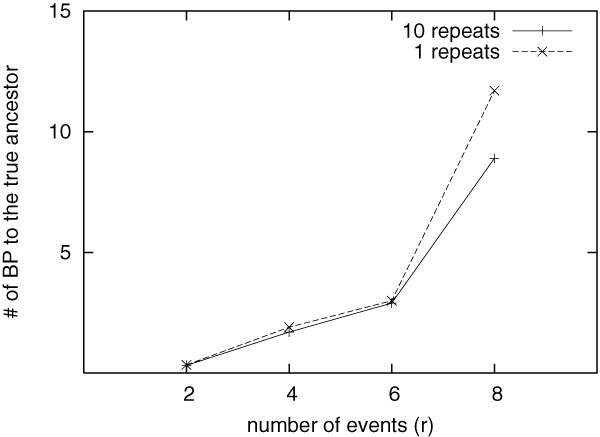
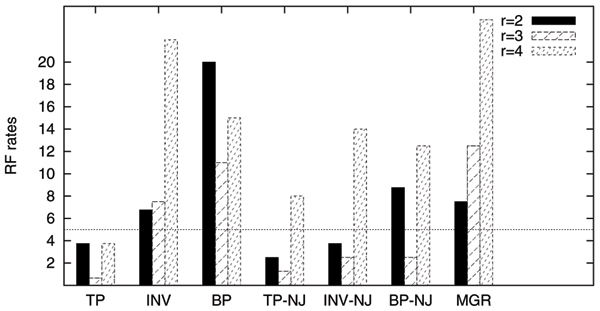
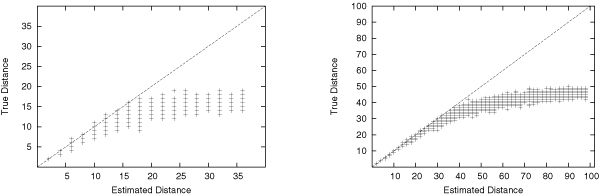
Our extensive testing using simulated datasets shows that GRAPPA-TP is very accurate in terms of ancestor genome inference and phylogenetic reconstruction. Simulation results also suggest that model match is critical in genome rearrangement analysis: it is not accurate to simulate transpositions with other events including inversions.
由于高通量测序技术的出现以及随之而来的测序成本降低,许多生物体已被完全测序,并且其大部分基因已被识别。因此,将整个基因组表示为基因标识符的有序列表并通过计算手段研究这些实体的重排成为可能。结果,基因组重排数据作为一种用于系统发育分析的新型数据,已引起生物学家和计算机科学家越来越多的关注。基因组重排的主要事件包括倒位、转座和颠换。迄今为止,GRAPPA和MGR是重排系统发育分析中最准确的方法,两者都假设倒位是唯一的事件。然而,由于计算转座距离的复杂性,当转座占主导时,很难分析数据集。
我们对GRAPPA进行了扩展以处理转座。新方法名为GRAPPA-TP,有两个主要扩展:一种估计转座距离的启发式方法,以及一种用于三个基因组的新的转座中位数求解器。虽然GRAPPA-TP使用贪心方法来计算转座距离,但当基因组相对接近时,它非常准确。新的GRAPPA-TP可从http://phylo.cse.sc.edu/获取。
我们使用模拟数据集进行的广泛测试表明,GRAPPA-TP在祖先基因组推断和系统发育重建方面非常准确。模拟结果还表明,模型匹配在基因组重排分析中至关重要:用包括倒位在内的其他事件来模拟转座是不准确的。