Gordon K L, Gonzalez-Alegre P
Interdisciplinary Graduate Program in Neuroscience, Carver College of Medicine, The University of Iowa, Iowa City, IA 52242, USA.
Neuroscience. 2008 Dec 2;157(3):588-95. doi: 10.1016/j.neuroscience.2008.09.028. Epub 2008 Sep 27.
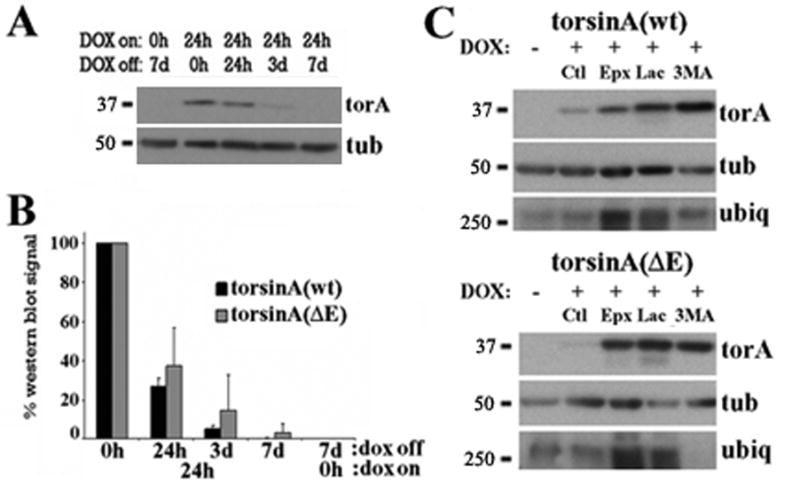
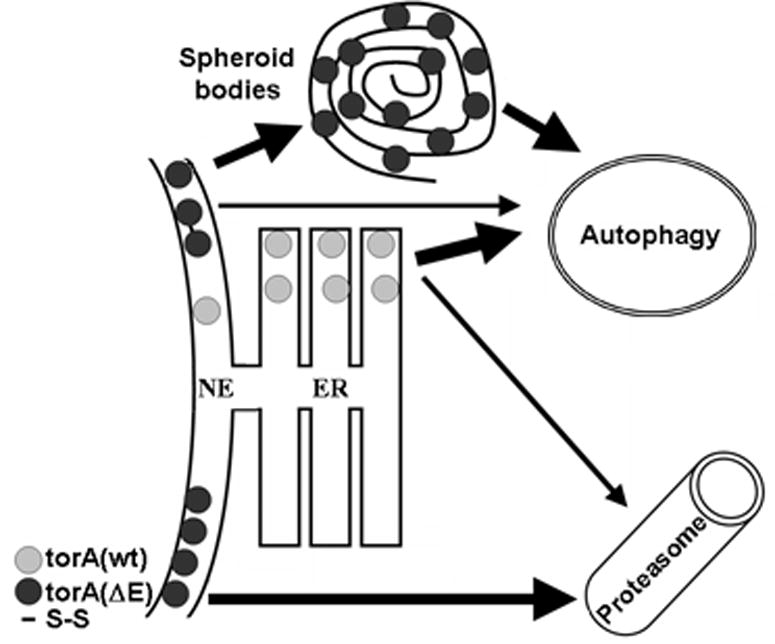
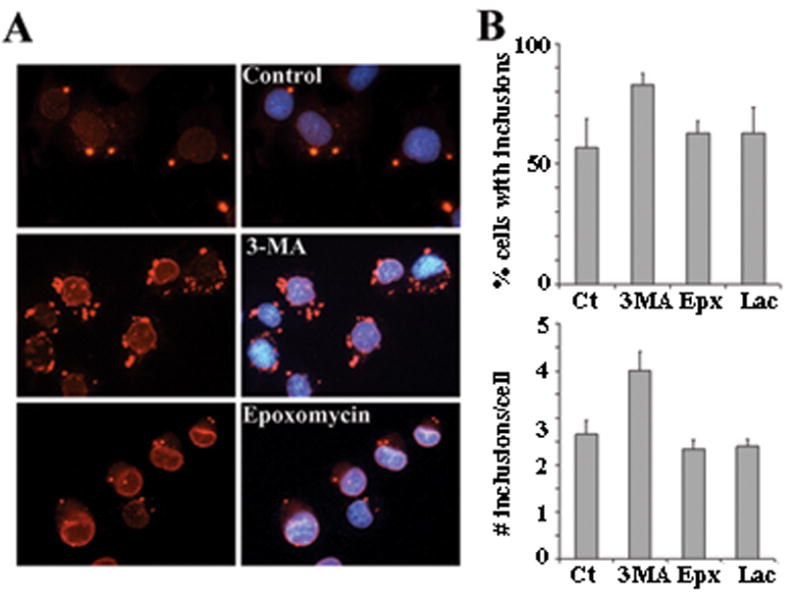
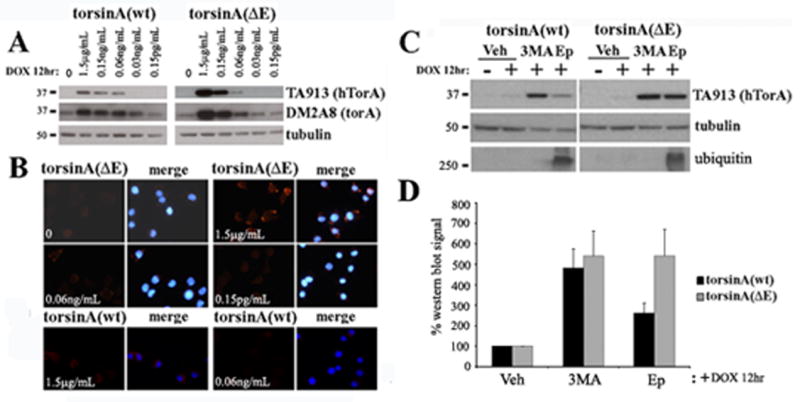
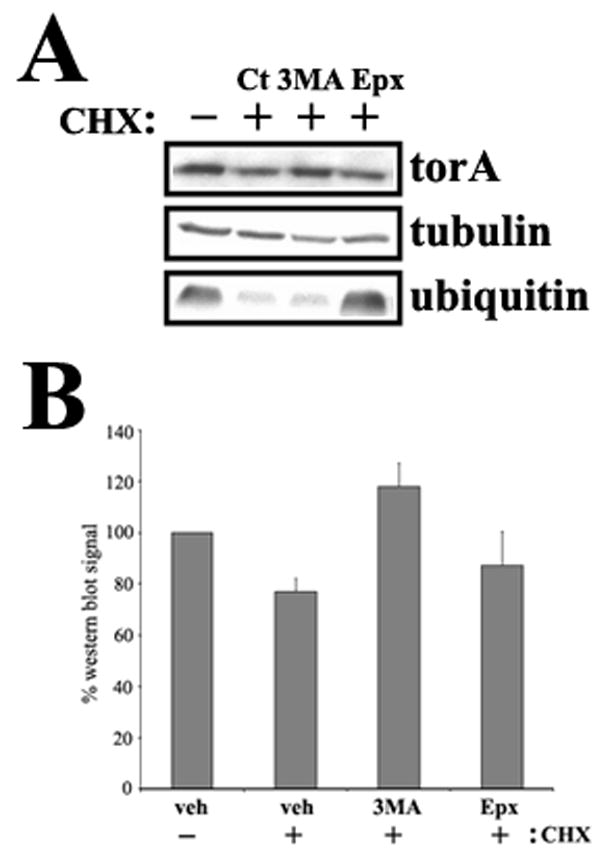
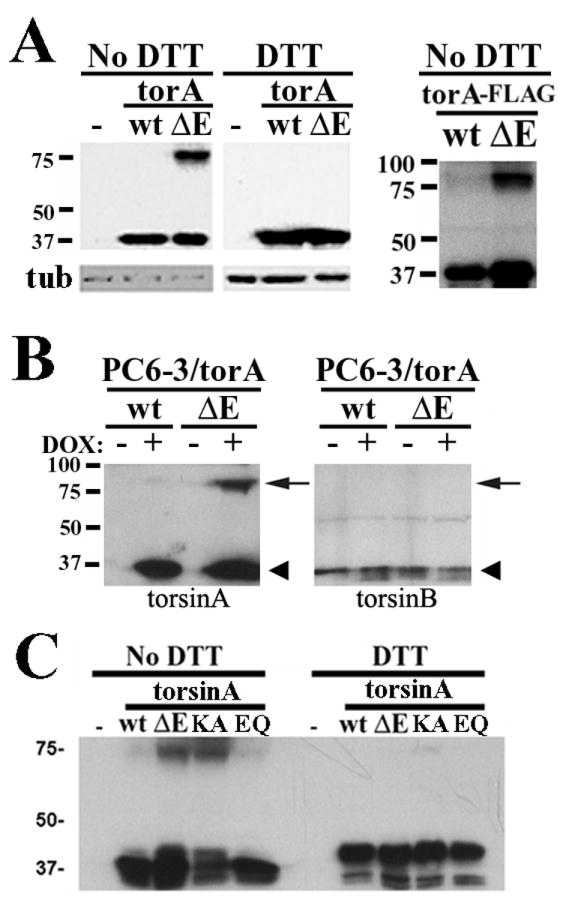
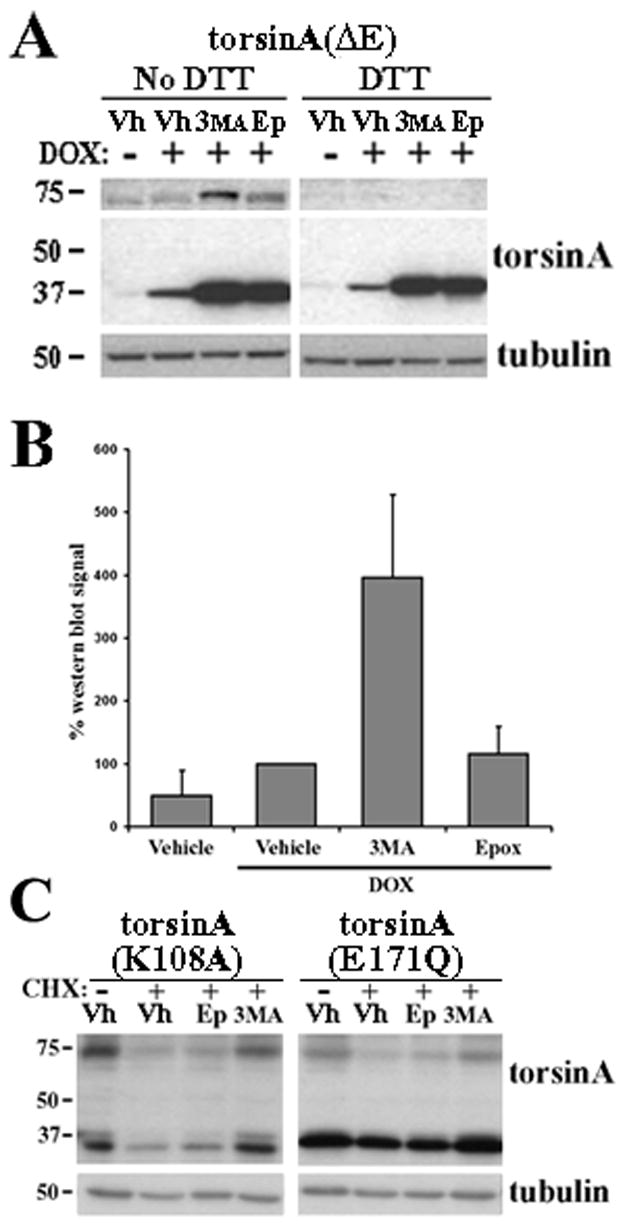
DYT1 is the most common inherited dystonia, a neurological syndrome that causes disabling involuntary muscle contractions. This autosomal dominant disease is caused by a glutamic acid deletion near the carboxy-terminus in the protein torsinA. Cell- and animal-based studies have shown how the DYT1 mutation causes mutant torsinA to redistribute from the endoplasmic reticulum to the nuclear envelope, acting through a dominant negative effect over the wild type protein. As a result, the wild type:mutant torsinA expression ratio would be important for disease pathogenesis, and events that influence it, such as a differential degradation process for each protein, might modulate DYT1 pathobiology. The DYT1 mutation also triggers the formation of abnormal intermolecular disulfide bonds in torsinA, although the significance of this finding is unclear. How the protein quality control machinery handles torsinA, and whether this process is affected by its abnormal oligomerization remain unknown. Here, we first explored how the disease-linked mutation influences the catabolic process of human torsinA, demonstrating that the differences in subcellular localization between both forms of torsinA lead to divergences in their degradation pathways and, whereas torsinA is normally recycled through autophagy, the proteasome is also required for the efficient clearance of the mutated form. Subsequently, we determined that the abnormal disulfide bond-dependent oligomerization of mutant torsinA is not a result of its redistribution to the nuclear envelope, but a direct consequence of the mutation. Finally, we established that the presence of disulfide links in mutant torsinA oligomers interfere with their degradation by the proteasome, thus relying on autophagy as the main pathway for clearance. In conclusion, the abnormal subcellular localization and oligomerization of DYT1-linked torsinA influences its catabolic process, opening the door to the modulation of the wild type:mutant torsinA ratio through pharmacological manipulation of protein degradation pathways.
DYT1是最常见的遗传性肌张力障碍,这是一种神经综合征,可导致使人丧失能力的非自主肌肉收缩。这种常染色体显性疾病是由torsinA蛋白羧基末端附近的谷氨酸缺失引起的。基于细胞和动物的研究已经表明DYT1突变是如何导致突变型torsinA从内质网重新分布到核膜,通过对野生型蛋白的显性负效应起作用。因此,野生型:突变型torsinA的表达比率对于疾病发病机制很重要,而影响该比率的事件,例如每种蛋白质的差异降解过程,可能会调节DYT1的病理生物学。DYT1突变还会触发torsinA中异常分子间二硫键的形成,尽管这一发现的意义尚不清楚。蛋白质质量控制机制如何处理torsinA,以及这一过程是否受到其异常寡聚化的影响仍然未知。在这里,我们首先探究了与疾病相关的突变如何影响人类torsinA的分解代谢过程,证明了两种形式的torsinA在亚细胞定位上的差异导致了它们降解途径的分歧,虽然torsinA通常通过自噬进行循环利用,但蛋白酶体对于有效清除突变形式也是必需的。随后,我们确定突变型torsinA异常的二硫键依赖性寡聚化不是其重新分布到核膜的结果,而是突变的直接后果。最后,我们证实突变型torsinA寡聚体中二硫键的存在会干扰它们被蛋白酶体降解,因此依赖自噬作为主要的清除途径。总之,与DYT1相关的torsinA异常的亚细胞定位和寡聚化影响其分解代谢过程,为通过对蛋白质降解途径进行药理学操作来调节野生型:突变型torsinA比率打开了大门。