Cruz Eugénia, Whittington Chris, Krikler Samuel H, Mascarenhas Cláudia, Lacerda Rosa, Vieira Jorge, Porto Graça
Clinical Hematology, Santo António Hospital, Porto, Portugal.
BMC Med Genet. 2008 Nov 6;9:97. doi: 10.1186/1471-2350-9-97.
Hereditary Hemochromatosis(HH) is a common genetic disorder of iron overload where the large majority of patients are homozygous for one ancestral mutation in the HFE gene. In spite of this remarkable genetic homogeneity, the condition is clinically heterogeneous, varying from a severe disease to an asymptomatic phenotype with only abnormal biochemical parameters. The recent recognition of the variable penetrance of the HH mutation in different large population studies demands the need to search for new modifiers of its phenotypic expression. The present study follows previous observations that MHC class-I linked genetic markers, associated with the setting of CD8+ T-lymphocyte numbers, could be clinically relevant modifiers of the phenotypic expression in HH, and aimed to find new markers that could be used as more reliable prognostic variables.
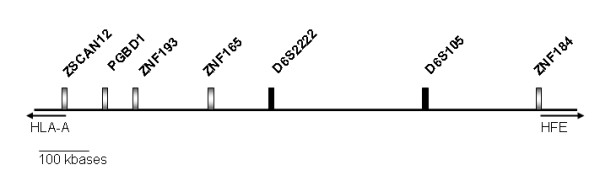
Haplotype analysis, including seven genetic markers within a 1 Mb region around the microsatellite D6S105 was performed in a group of 56 previously characterized C282Y homozygous Portuguese patients. Parameters analyzed in this study were total body iron stores, clinical manifestations related with HH and immunological parameters (total lymphocyte numbers, CD4+ and CD8+ T-lymphocyte numbers). An independent group of 10 C282Y homozygous patients from Vancouver, Canada, were also included in this study and analyzed for the same parameters.
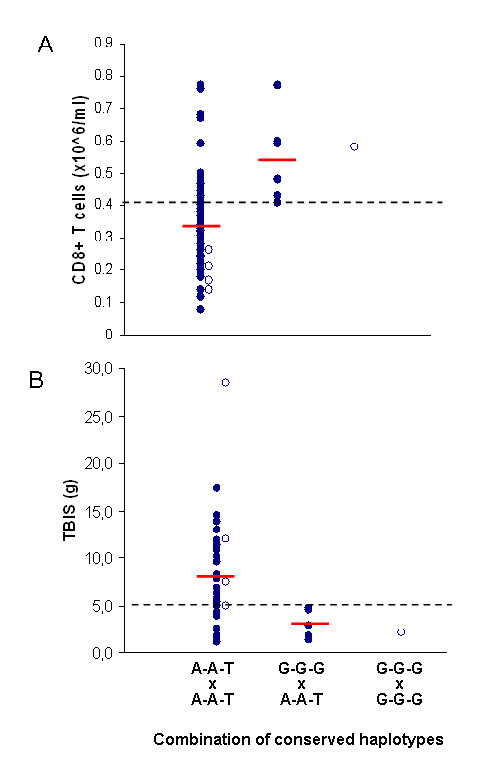
A highly conserved ancestral haplotype defined by the SNP markers PGBD1-A, ZNF193-A, ZNF165-T (designated as A-A-T) was found associated with both abnormally low CD8+ T-lymphocyte numbers and the development of a severe clinical expression of HH. In a small proportion of patients, another conserved haplotype defined by the SNP markers PGBD1-G, ZNF193-G, ZNF165-G (designated as G-G-G) was found associated with high CD8+ T-lymphocyte numbers and a milder clinical expression. Remarkably, the two conserved haplotypes defined in Portuguese patients were also observed in the geographically different population of Canadian patients, also predicting CD8+ T-lymphocyte numbers and the severity of disease.
These results may have important implications not only for approaching the question of the penetrance of the hemochromatosis gene in different world populations but also to further narrow the region of interest to find a candidate gene involved in the setting of CD8+ T-lymphocyte numbers in humans.
遗传性血色素沉着症(HH)是一种常见的铁过载遗传疾病,绝大多数患者为HFE基因一个祖先突变的纯合子。尽管存在这种显著的遗传同质性,但该病症在临床上具有异质性,从严重疾病到仅生化参数异常的无症状表型不等。最近在不同大规模人群研究中对HH突变可变外显率的认识,要求寻找其表型表达的新修饰因子。本研究遵循先前的观察结果,即与CD8 + T淋巴细胞数量设定相关的MHC I类连锁遗传标记可能是HH表型表达的临床相关修饰因子,旨在寻找可作为更可靠预后变量的新标记。
对一组56名先前已鉴定的C282Y纯合葡萄牙患者进行单倍型分析,包括微卫星D6S105周围1 Mb区域内的7个遗传标记。本研究分析的参数包括全身铁储存、与HH相关的临床表现和免疫参数(总淋巴细胞数量、CD4 +和CD8 + T淋巴细胞数量)。来自加拿大温哥华的10名C282Y纯合患者的独立组也纳入本研究,并对相同参数进行分析。
发现由单核苷酸多态性(SNP)标记PGBD1 - A、ZNF193 - A、ZNF165 - T定义的高度保守祖先单倍型(称为A - A - T)与异常低的CD8 + T淋巴细胞数量以及HH的严重临床表型发展相关。在一小部分患者中,发现由SNP标记PGBD1 - G、ZNF193 - G、ZNF165 - G定义的另一种保守单倍型(称为G - G - G)与高CD8 + T淋巴细胞数量和较轻的临床表型相关。值得注意的是,在地理上不同的加拿大患者群体中也观察到了在葡萄牙患者中定义的两种保守单倍型,它们同样可预测CD8 + T淋巴细胞数量和疾病严重程度。
这些结果不仅可能对解决血色素沉着症基因在不同世界人群中的外显率问题具有重要意义,而且对于进一步缩小感兴趣区域以找到参与人类CD8 + T淋巴细胞数量设定的候选基因也具有重要意义。