Molecular Pathology Research and Development Laboratory, Department of Pathology, Peter MacCallum Cancer Centre, Melbourne, Victoria 8006, Australia.
Epigenetics Chromatin. 2008 Nov 3;1(1):7. doi: 10.1186/1756-8935-1-7.
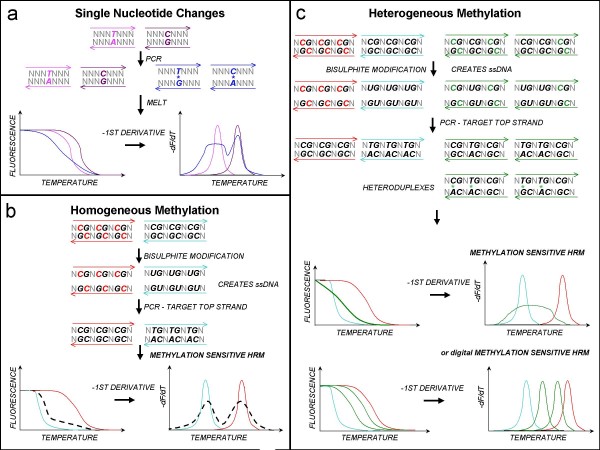
Methylation-sensitive high resolution melting (MS-HRM) methodology is able to recognise heterogeneously methylated sequences by their characteristic melting profiles. To further analyse heterogeneously methylated sequences, we adopted a digital approach to MS-HRM (dMS-HRM) that involves the amplification of single templates after limiting dilution to quantify and to determine the degree of methylation. We used this approach to study methylation of the CDKN2B (p15) cell cycle progression inhibitor gene which is inactivated by DNA methylation in haematological malignancies of the myeloid lineage. Its promoter region usually shows heterogeneous methylation and is only rarely fully methylated. The methylation status of CDKN2B can be used as a biomarker of response to treatment. Therefore the accurate characterisation of its methylation is desirable.
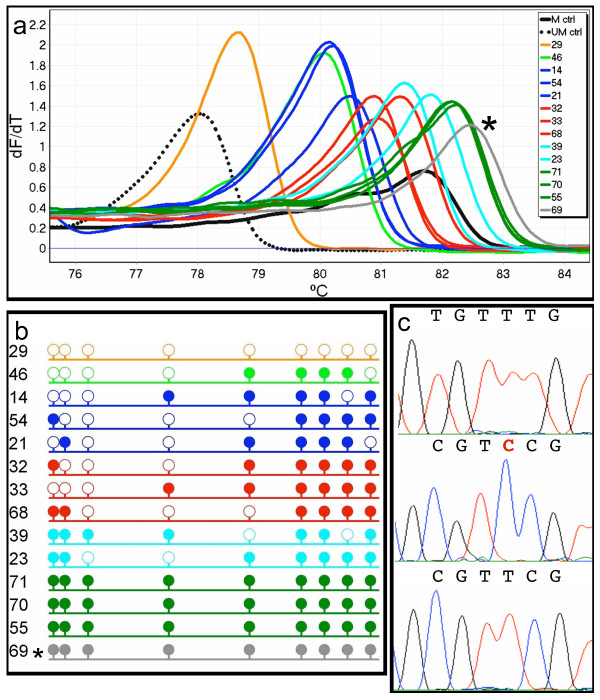
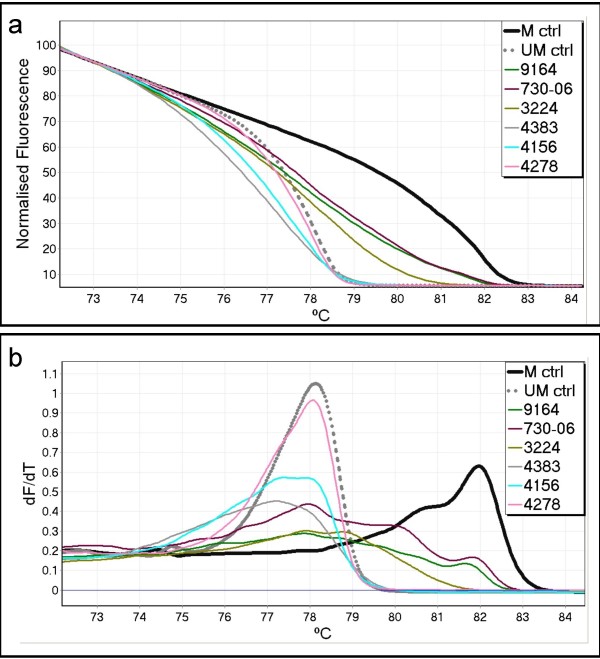
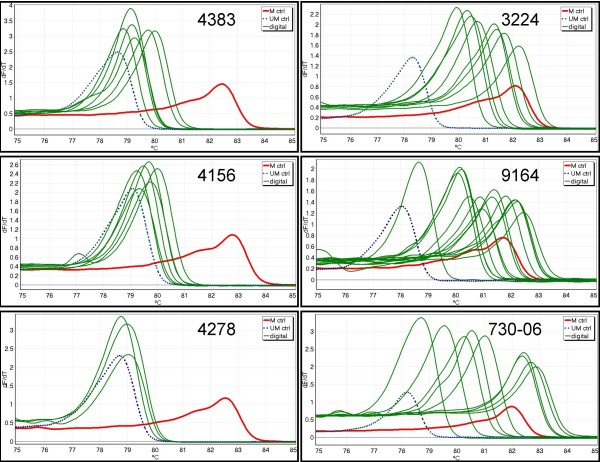
MS-HRM was used to assess CDKN2B methylation in acute myeloid leukaemia (AML) samples. All the AML samples that were methylated at the CDKN2B promoter (40/93) showed varying degrees of heterogeneous methylation. Six representative samples were selected for further study. dMS-HRM was used to simultaneously count the methylated alleles and assess the degree of methylation. Direct sequencing of selected dMS-HRM products was used to determine the exact DNA methylation pattern and confirmed the degree of methylation estimated by dMS-HRM.
dMS-HRM is a powerful technique for the analysis of methylation in CDKN2B and other heterogeneously methylated genes. It eliminates both PCR and cloning bias towards either methylated or unmethylated DNA. Potentially complex information is simplified into a digital output, allowing counting of methylated and unmethylated alleles and providing an overall picture of methylation at the given locus. Downstream sequencing is minimised as dMS-HRM acts as a screen to select only methylated clones for further analysis.
甲基化敏感高分辨率熔解(MS-HRM)方法能够通过其特征熔解曲线识别异甲基化序列。为了进一步分析异甲基化序列,我们采用了数字 MS-HRM(dMS-HRM)方法,该方法涉及在限制稀释后扩增单个模板,以定量和确定甲基化程度。我们使用这种方法来研究 CDKN2B(p15)细胞周期抑制剂基因的甲基化,该基因在髓系血液恶性肿瘤中因 DNA 甲基化而失活。其启动子区域通常表现出异甲基化,很少完全甲基化。CDKN2B 的甲基化状态可用作治疗反应的生物标志物。因此,准确描述其甲基化状态是可取的。
MS-HRM 用于评估急性髓细胞白血病(AML)样本中 CDKN2B 的甲基化。所有 CDKN2B 启动子甲基化的 AML 样本(40/93)均显示出不同程度的异甲基化。选择了六个有代表性的样本进行进一步研究。dMS-HRM 用于同时计数甲基化等位基因并评估甲基化程度。选择的 dMS-HRM 产物的直接测序用于确定确切的 DNA 甲基化模式,并证实了 dMS-HRM 估计的甲基化程度。
dMS-HRM 是分析 CDKN2B 和其他异甲基化基因甲基化的强大技术。它消除了 PCR 和克隆对甲基化或未甲基化 DNA 的偏向。潜在的复杂信息被简化为数字输出,允许计数甲基化和未甲基化等位基因,并提供给定基因座甲基化的总体情况。由于 dMS-HRM 可以作为筛选,仅选择进一步分析的甲基化克隆,因此下游测序被最小化。