Peto Myron, Kloczkowski Andrzej, Honavar Vasant, Jernigan Robert L
Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, IA 50011-3020, USA.
BMC Bioinformatics. 2008 Nov 18;9:487. doi: 10.1186/1471-2105-9-487.
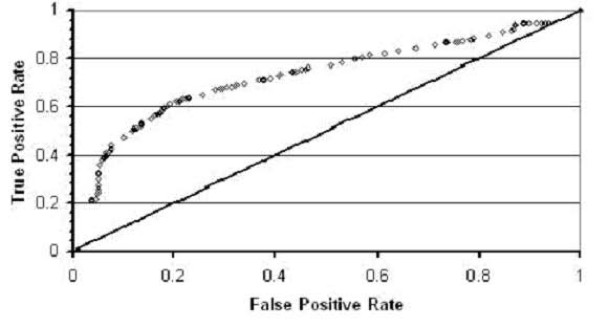
By using a standard Support Vector Machine (SVM) with a Sequential Minimal Optimization (SMO) method of training, Naïve Bayes and other machine learning algorithms we are able to distinguish between two classes of protein sequences: those folding to highly-designable conformations, or those folding to poorly- or non-designable conformations.
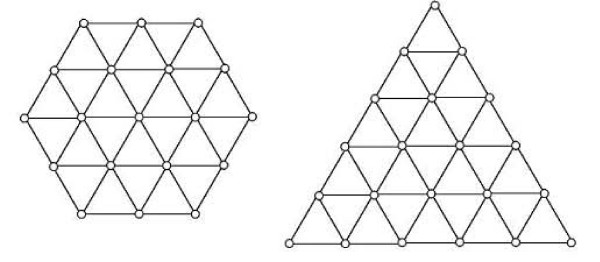
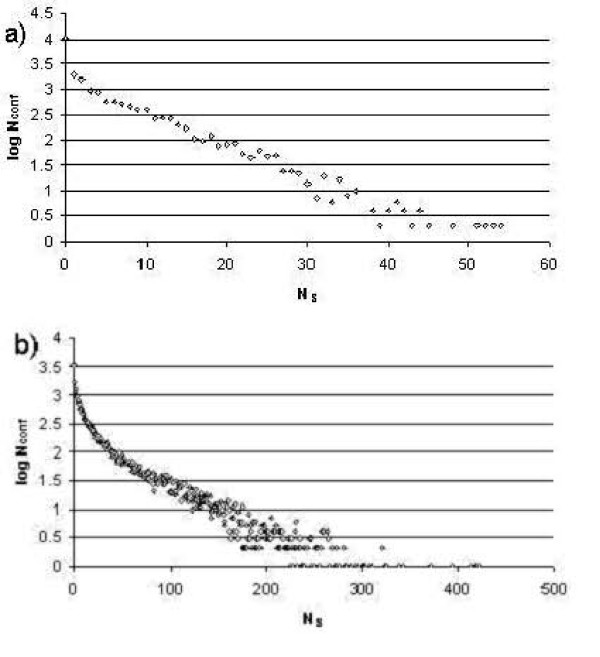
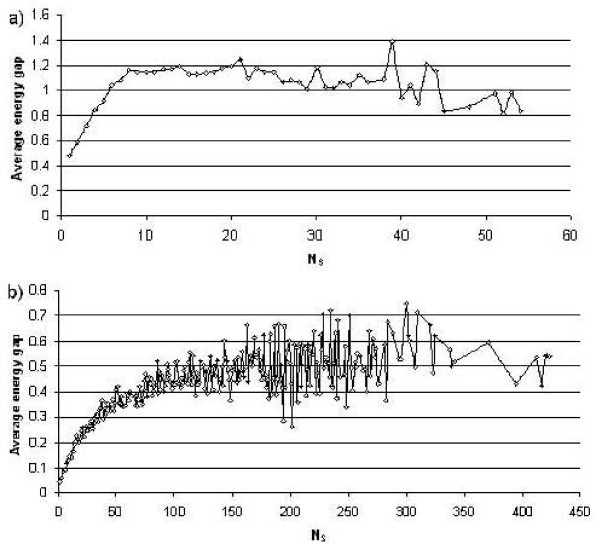
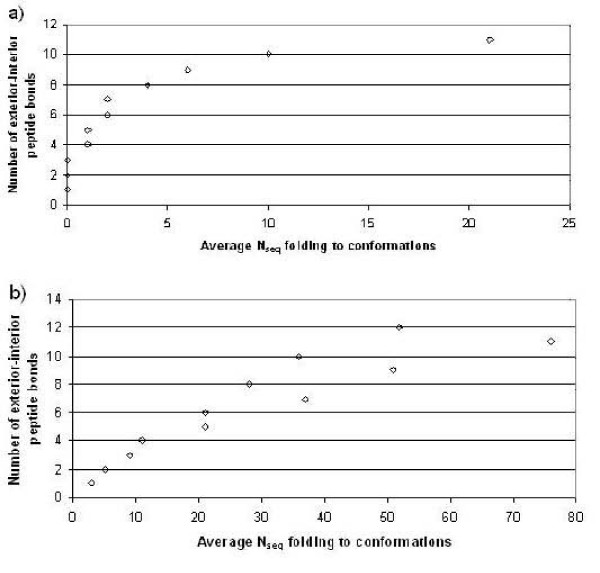
First, we generate all possible compact lattice conformations for the specified shape (a hexagon or a triangle) on the 2D triangular lattice. Then we generate all possible binary hydrophobic/polar (H/P) sequences and by using a specified energy function, thread them through all of these compact conformations. If for a given sequence the lowest energy is obtained for a particular lattice conformation we assume that this sequence folds to that conformation. Highly-designable conformations have many H/P sequences folding to them, while poorly-designable conformations have few or no H/P sequences. We classify sequences as folding to either highly- or poorly-designable conformations. We have randomly selected subsets of the sequences belonging to highly-designable and poorly-designable conformations and used them to train several different standard machine learning algorithms.
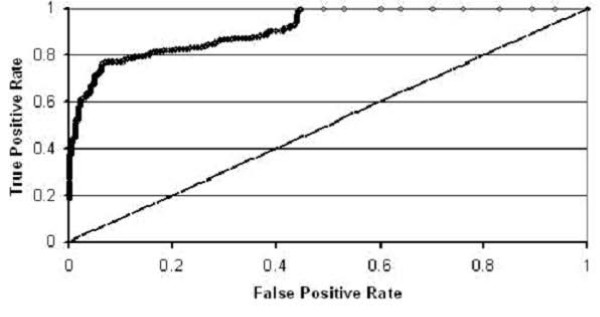
By using these machine learning algorithms with ten-fold cross-validation we are able to classify the two classes of sequences with high accuracy -- in some cases exceeding 95%.
通过使用带有序列最小优化(SMO)训练方法的标准支持向量机(SVM)、朴素贝叶斯和其他机器学习算法,我们能够区分两类蛋白质序列:那些折叠成高度可设计构象的序列,以及那些折叠成低可设计或不可设计构象的序列。
首先,我们在二维三角形晶格上为指定形状(六边形或三角形)生成所有可能的紧凑晶格构象。然后我们生成所有可能的二元疏水/极性(H/P)序列,并使用指定的能量函数,将它们穿入所有这些紧凑构象中。如果对于给定序列,特定晶格构象获得最低能量,我们就假设该序列折叠成该构象。高度可设计的构象有许多H/P序列折叠到它们上面,而低可设计的构象则很少或没有H/P序列。我们将序列分类为折叠成高度可设计或低可设计构象。我们随机选择了属于高度可设计和低可设计构象的序列子集,并使用它们来训练几种不同的标准机器学习算法。
通过使用这些具有十折交叉验证的机器学习算法,我们能够以高精度对这两类序列进行分类——在某些情况下超过95%。