Omont Nicolas, Forner Karl, Lamarine Marc, Martin Gwendal, Képès François, Wojcik Jérôme
Merck Serono International S,A, 9 chemin des Mines, 1202 Geneva, Switzerland.
BMC Proc. 2008 Dec 17;2 Suppl 4(Suppl 4):S6. doi: 10.1186/1753-6561-2-s4-s6.
With the improvement of genotyping technologies and the exponentially growing number of available markers, case-control genome-wide association studies promise to be a key tool for investigation of complex diseases. However new analytical methods have to be developed to face the problems induced by this data scale-up, such as statistical multiple testing, data quality control and computational tractability.
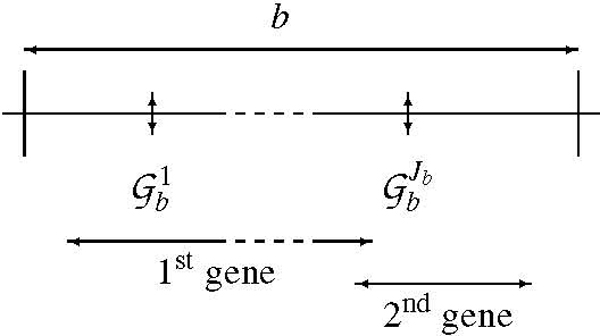
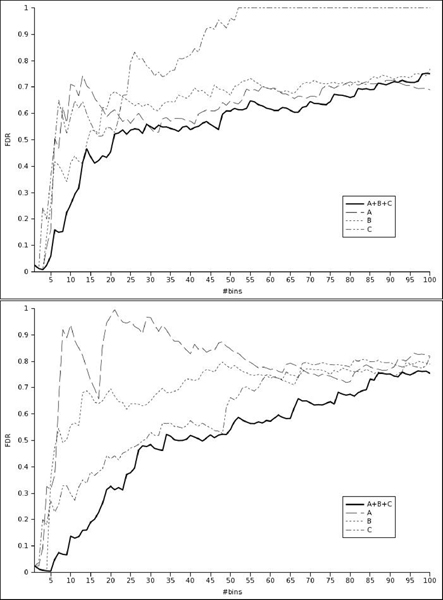
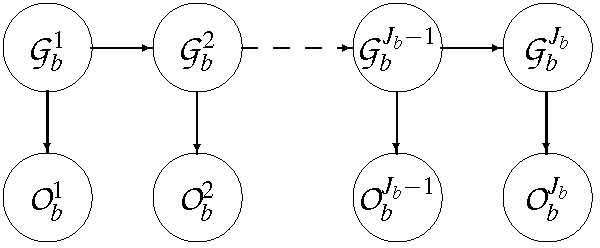
We present a novel method to analyze genome-wide association studies results. The algorithm is based on a Bayesian model that integrates genotyping errors and genomic structure dependencies. p-values are assigned to genomic regions termed bins, which are defined from a gene-biased partitioning of the genome, and the false-discovery rate is estimated. We have applied this algorithm to data coming from three genome-wide association studies of Multiple Sclerosis.
The method practically overcomes the scale-up problems and permits to identify new putative regions statistically associated with the disease.
随着基因分型技术的改进以及可用标记数量呈指数级增长,病例对照全基因组关联研究有望成为研究复杂疾病的关键工具。然而,必须开发新的分析方法来应对这种数据规模扩大所带来的问题,例如统计多重检验、数据质量控制和计算可处理性。
我们提出了一种分析全基因组关联研究结果的新方法。该算法基于一个整合了基因分型错误和基因组结构依赖性的贝叶斯模型。将p值分配给称为“bin”的基因组区域,这些区域是根据基因组的基因偏向划分定义的,并估计错误发现率。我们已将此算法应用于来自三项多发性硬化症全基因组关联研究的数据。
该方法切实克服了规模扩大问题,并能够识别出与疾病有统计学关联的新的假定区域。