Parry David A, Mighell Alan J, El-Sayed Walid, Shore Roger C, Jalili Ismail K, Dollfus Hélène, Bloch-Zupan Agnes, Carlos Roman, Carr Ian M, Downey Louise M, Blain Katharine M, Mansfield David C, Shahrabi Mehdi, Heidari Mansour, Aref Parissa, Abbasi Mohsen, Michaelides Michel, Moore Anthony T, Kirkham Jennifer, Inglehearn Chris F
Leeds Institute of Molecular Medicine, University of Leeds, St. James's University Hospital, Leeds LS9 7TF, UK.
Am J Hum Genet. 2009 Feb;84(2):266-73. doi: 10.1016/j.ajhg.2009.01.009. Epub 2009 Feb 5.
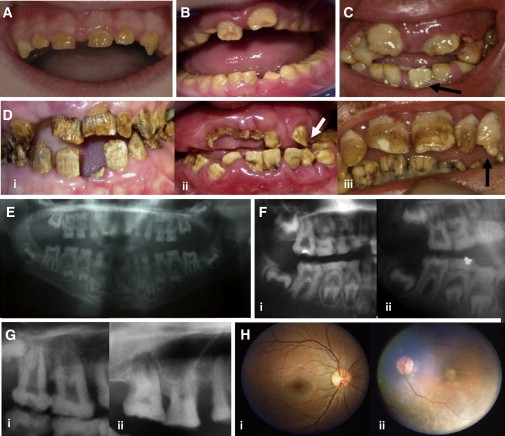
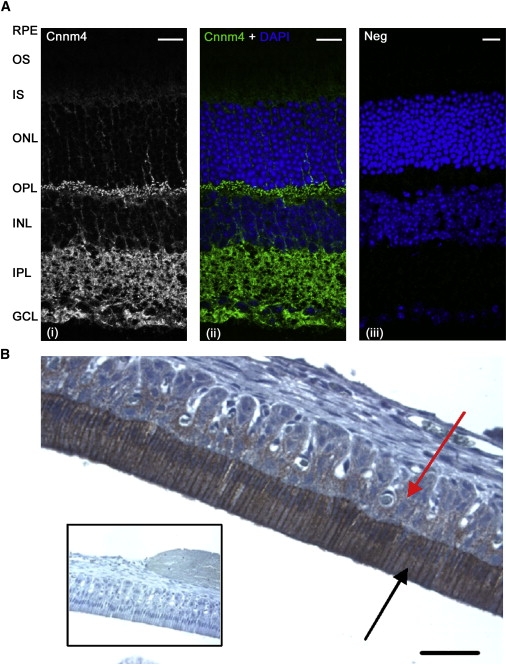
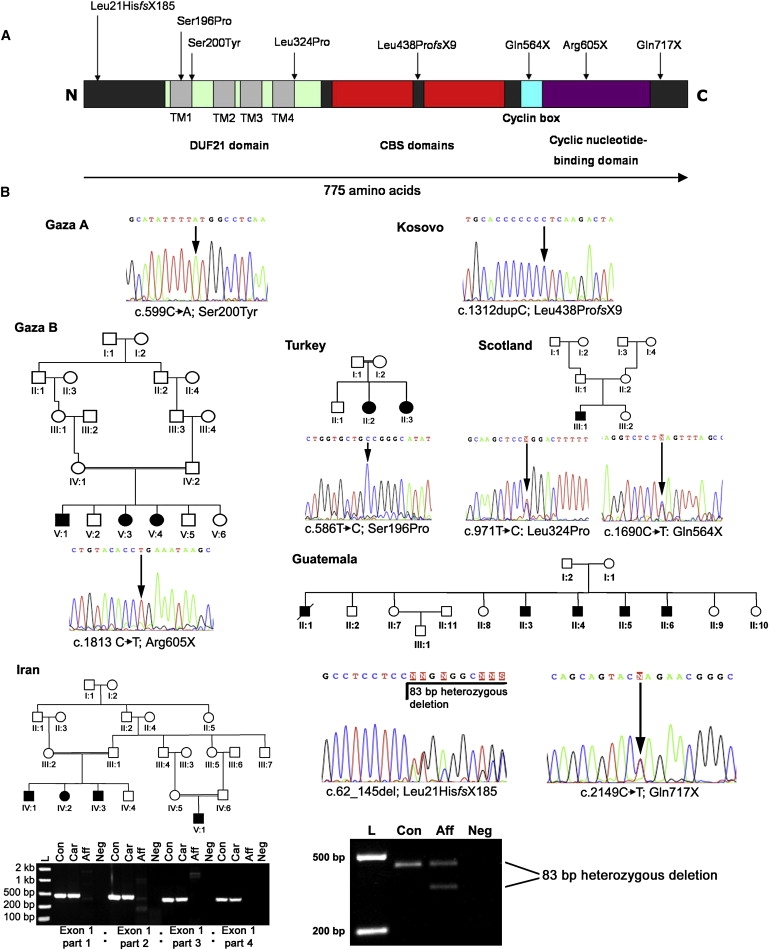
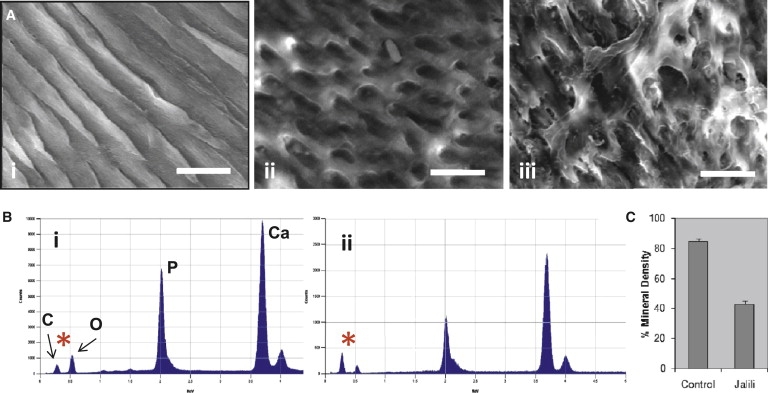
The combination of recessively inherited cone-rod dystrophy (CRD) and amelogenesis imperfecta (AI) was first reported by Jalili and Smith in 1988 in a family subsequently linked to a locus on chromosome 2q11, and it has since been reported in a second small family. We have identified five further ethnically diverse families cosegregating CRD and AI. Phenotypic characterization of teeth and visual function in the published and new families reveals a consistent syndrome in all seven families, and all link or are consistent with linkage to 2q11, confirming the existence of a genetically homogenous condition that we now propose to call Jalili syndrome. Using a positional-candidate approach, we have identified mutations in the CNNM4 gene, encoding a putative metal transporter, accounting for the condition in all seven families. Nine mutations are described in all, three missense, three terminations, two large deletions, and a single base insertion. We confirmed expression of Cnnm4 in the neural retina and in ameloblasts in the developing tooth, suggesting a hitherto unknown connection between tooth biomineralization and retinal function. The identification of CNNM4 as the causative gene for Jalili syndrome, characterized by syndromic CRD with AI, has the potential to provide new insights into the roles of metal transport in visual function and biomineralization.
隐性遗传的视锥-视杆营养不良(CRD)与牙釉质发育不全(AI)的联合病症于1988年由贾利利和史密斯首次报道,该家族随后被定位到2号染色体q11位点,此后在另一个小家族中也有报道。我们又鉴定出另外五个种族不同的家族,其CRD和AI共分离。已发表家族和新家族中牙齿和视觉功能的表型特征显示,所有七个家族都存在一种一致的综合征,且所有家族均与2q11连锁或符合连锁关系,证实存在一种我们现在提议称为贾利利综合征的基因同质病症。通过位置候选方法,我们在编码一种假定金属转运蛋白的CNNM4基因中鉴定出突变,该突变解释了所有七个家族的病症。总共描述了九个突变,三个错义突变、三个终止突变、两个大的缺失和一个单碱基插入。我们证实了Cnnm4在神经视网膜和发育中牙齿的成釉细胞中的表达,这表明牙齿生物矿化与视网膜功能之间存在迄今未知的联系。将CNNM4鉴定为以伴有AI的综合征性CRD为特征的贾利利综合征的致病基因,有可能为金属转运在视觉功能和生物矿化中的作用提供新的见解。