Stark Zornitza, Savarirayan Ravi
Genetic Health Services Victoria, and Murdoch Childrens Research Institute, Melbourne, Australia.
Orphanet J Rare Dis. 2009 Feb 20;4:5. doi: 10.1186/1750-1172-4-5.
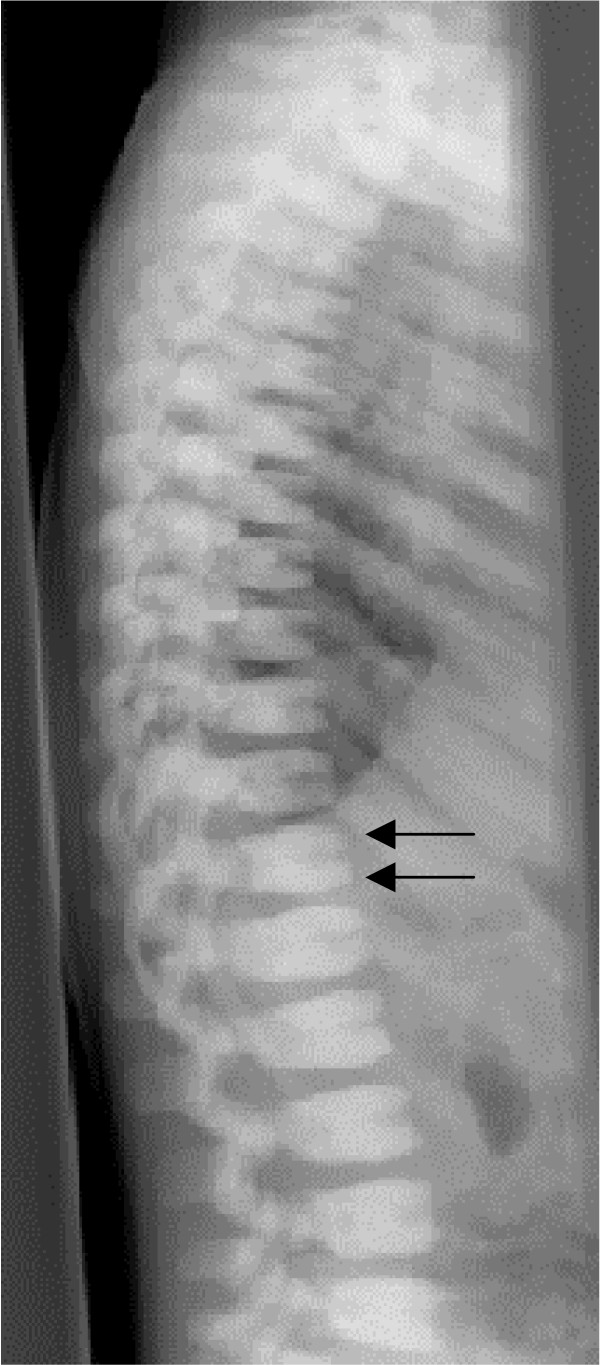
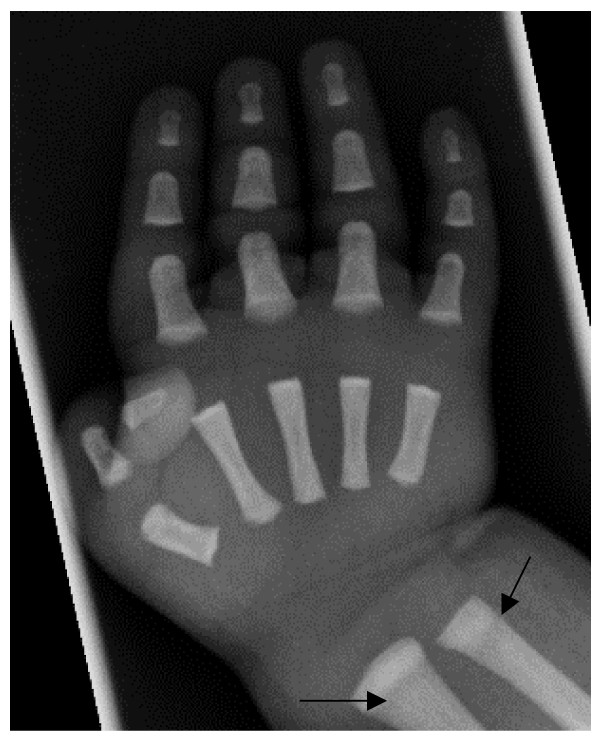
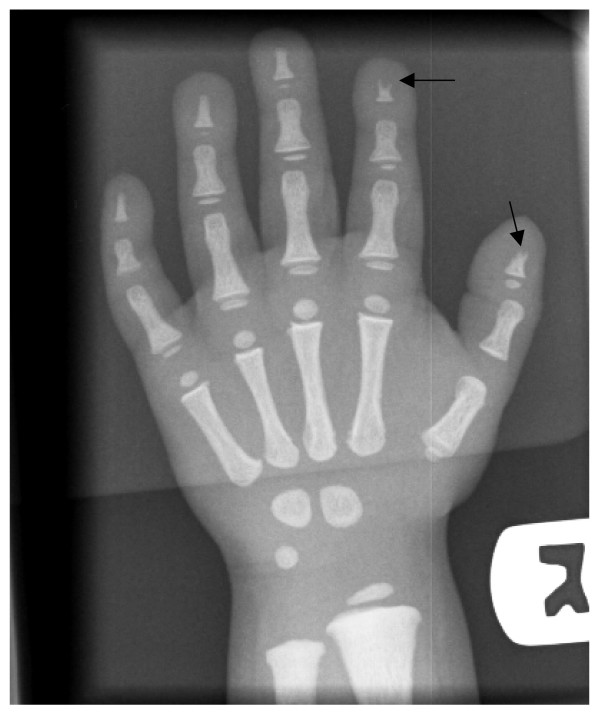
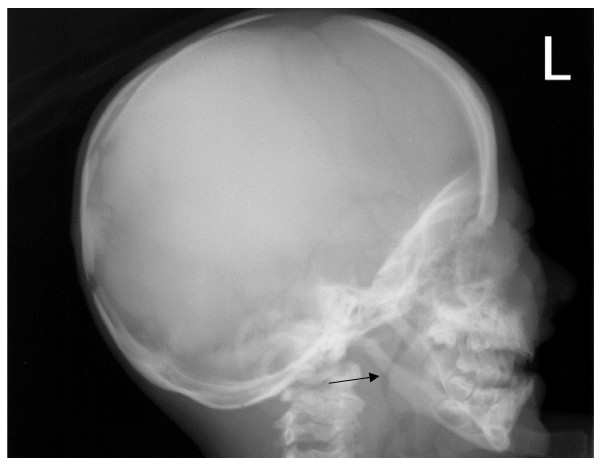
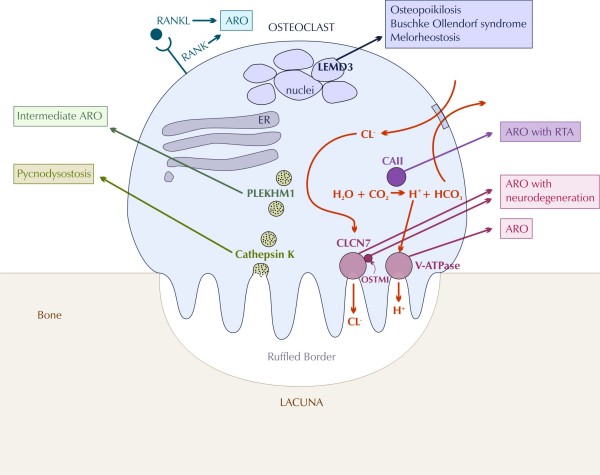
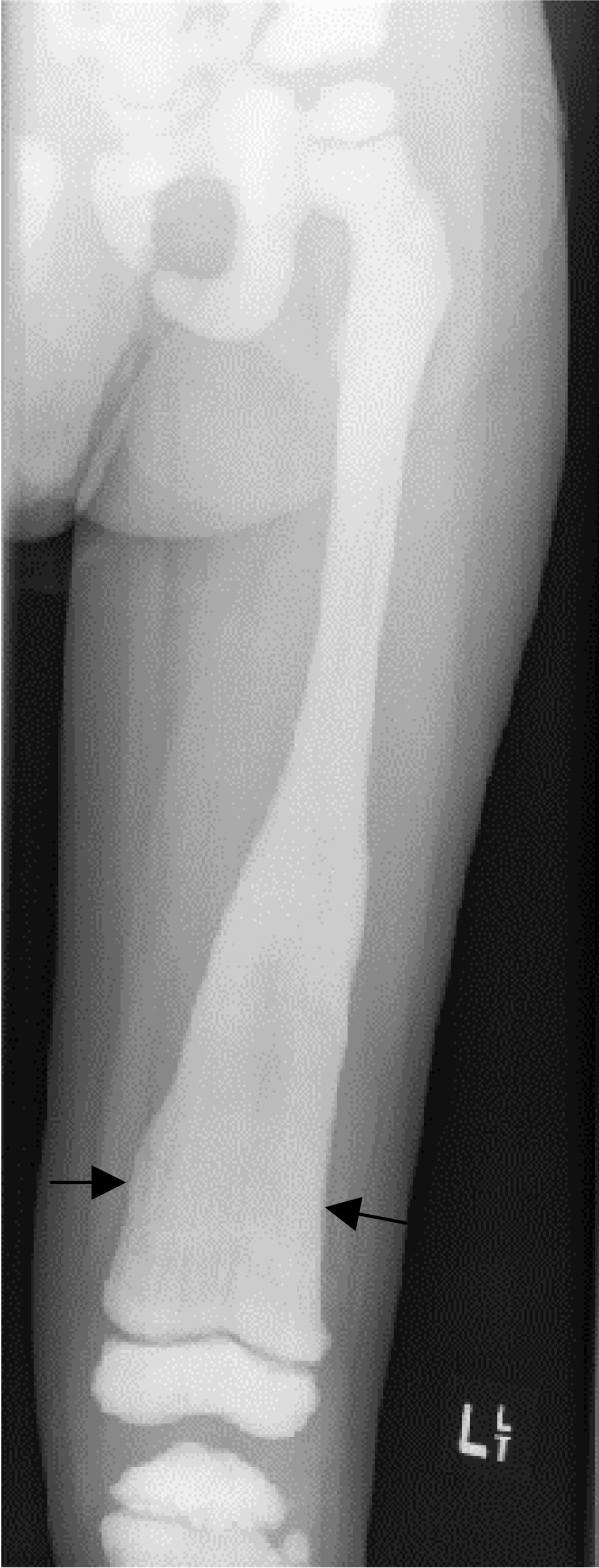
Osteopetrosis ("marble bone disease") is a descriptive term that refers to a group of rare, heritable disorders of the skeleton characterized by increased bone density on radiographs. The overall incidence of these conditions is difficult to estimate but autosomal recessive osteopetrosis (ARO) has an incidence of 1 in 250,000 births, and autosomal dominant osteopetrosis (ADO) has an incidence of 1 in 20,000 births. Osteopetrotic conditions vary greatly in their presentation and severity, ranging from neonatal onset with life-threatening complications such as bone marrow failure (e.g. classic or "malignant" ARO), to the incidental finding of osteopetrosis on radiographs (e.g. osteopoikilosis). Classic ARO is characterised by fractures, short stature, compressive neuropathies, hypocalcaemia with attendant tetanic seizures, and life-threatening pancytopaenia. The presence of primary neurodegeneration, mental retardation, skin and immune system involvement, or renal tubular acidosis may point to rarer osteopetrosis variants, whereas onset of primarily skeletal manifestations such as fractures and osteomyelitis in late childhood or adolescence is typical of ADO. Osteopetrosis is caused by failure of osteoclast development or function and mutations in at least 10 genes have been identified as causative in humans, accounting for 70% of all cases. These conditions can be inherited as autosomal recessive, dominant or X-linked traits with the most severe forms being autosomal recessive. Diagnosis is largely based on clinical and radiographic evaluation, confirmed by gene testing where applicable, and paves the way to understanding natural history, specific treatment where available, counselling regarding recurrence risks, and prenatal diagnosis in severe forms. Treatment of osteopetrotic conditions is largely symptomatic, although haematopoietic stem cell transplantation is employed for the most severe forms associated with bone marrow failure and currently offers the best chance of longer-term survival in this group. The severe infantile forms of osteopetrosis are associated with diminished life expectancy, with most untreated children dying in the first decade as a complication of bone marrow suppression. Life expectancy in the adult onset forms is normal. It is anticipated that further understanding of the molecular pathogenesis of these conditions will reveal new targets for pharmacotherapy.
骨质石化症(“大理石骨病”)是一个描述性术语,指的是一组罕见的遗传性骨骼疾病,其特征是在X线片上骨密度增加。这些病症的总体发病率难以估计,但常染色体隐性遗传性骨质石化症(ARO)的发病率为每250,000例出生中有1例,常染色体显性遗传性骨质石化症(ADO)的发病率为每20,000例出生中有1例。骨质石化症的临床表现和严重程度差异很大,从新生儿期发病并伴有危及生命的并发症,如骨髓衰竭(如典型或“恶性”ARO),到X线片上偶然发现骨质石化症(如骨斑点症)。典型的ARO表现为骨折、身材矮小、压迫性神经病变、低钙血症伴手足抽搐发作,以及危及生命的全血细胞减少。存在原发性神经变性、智力发育迟缓、皮肤和免疫系统受累或肾小管酸中毒可能提示罕见的骨质石化症变体,而在儿童晚期或青少年期主要出现骨骼表现,如骨折和骨髓炎,则是ADO的典型特征。骨质石化症是由破骨细胞发育或功能障碍引起的,已确定至少10个基因的突变是人类致病原因,占所有病例的70%。这些病症可作为常染色体隐性、显性或X连锁性状遗传,最严重的形式为常染色体隐性遗传。诊断主要基于临床和影像学评估,在适用时通过基因检测确诊,并为了解自然病史、提供可用的特定治疗、咨询复发风险以及对严重形式进行产前诊断铺平道路。骨质石化症的治疗主要是对症治疗,尽管造血干细胞移植用于与骨髓衰竭相关的最严重形式,目前为该组患者提供了最佳的长期生存机会。严重的婴儿型骨质石化症与预期寿命缩短有关,大多数未经治疗的儿童在第一个十年内死于骨髓抑制并发症。成人发病形式的预期寿命正常。预计对这些病症分子发病机制的进一步了解将揭示药物治疗的新靶点。