Feng Chen, Putonti Catherine, Zhang Meizhuo, Eggers Rick, Mitra Rahul, Hogan Mike, Jayaraman Krishna, Fofanov Yuriy
Department of Computer Science, University of Houston, Houston, TX, USA.
BMC Genomics. 2009 Feb 20;10:85. doi: 10.1186/1471-2164-10-85.
The variations within an individual's HLA (Human Leukocyte Antigen) genes have been linked to many immunological events, e.g. susceptibility to disease, response to vaccines, and the success of blood, tissue, and organ transplants. Although the microarray format has the potential to achieve high-resolution typing, this has yet to be attained due to inefficiencies of current probe design strategies.
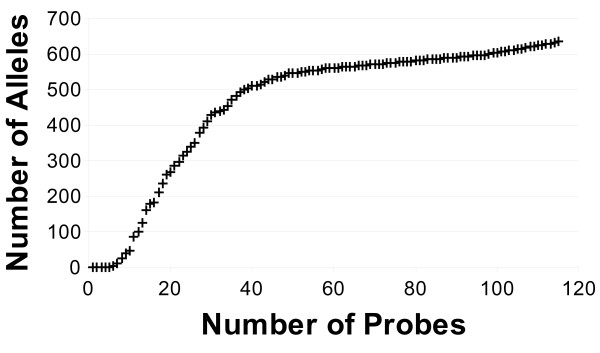
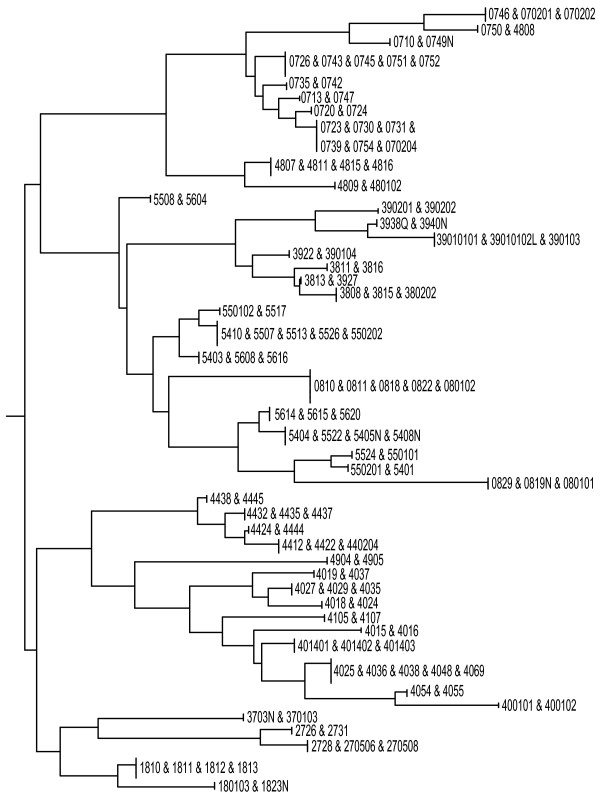
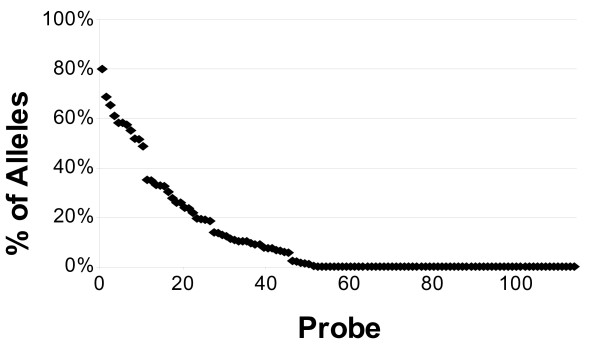
We present a novel three-step approach for the design of high-throughput microarray assays for HLA typing. This approach first selects sequences containing the SNPs present in all alleles of the locus of interest and next calculates the number of base changes necessary to convert a candidate probe sequences to the closest subsequence within the set of sequences that are likely to be present in the sample including the remainder of the human genome in order to identify those candidate probes which are "ultraspecific" for the allele of interest. Due to the high specificity of these sequences, it is possible that preliminary steps such as PCR amplification are no longer necessary. Lastly, the minimum number of these ultraspecific probes is selected such that the highest resolution typing can be achieved for the minimal cost of production. As an example, an array was designed and in silico results were obtained for typing of the HLA-B locus.
The assay presented here provides a higher resolution than has previously been developed and includes more alleles than previously considered. Based upon the in silico and preliminary experimental results, we believe that the proposed approach can be readily applied to any highly polymorphic gene system.
个体的人类白细胞抗原(HLA)基因变异与许多免疫事件相关,例如疾病易感性、疫苗反应以及血液、组织和器官移植的成功率。尽管微阵列形式有潜力实现高分辨率分型,但由于当前探针设计策略的低效性,尚未达到这一目标。
我们提出了一种用于HLA分型的高通量微阵列检测设计的新颖三步法。该方法首先选择包含感兴趣位点所有等位基因中存在的单核苷酸多态性(SNP)的序列,然后计算将候选探针序列转换为样本中可能存在的序列集合(包括人类基因组其余部分)内最接近的子序列所需的碱基变化数量,以便识别那些对感兴趣等位基因“超特异性”的候选探针。由于这些序列的高特异性,可能不再需要诸如聚合酶链反应(PCR)扩增等初步步骤。最后,选择这些超特异性探针的最小数量,以便以最低的生产成本实现最高分辨率的分型。例如,设计了一个阵列,并获得了用于HLA - B位点分型的计算机模拟结果。
本文提出的检测方法提供了比以前开发的方法更高的分辨率,并且包含比以前考虑的更多的等位基因。基于计算机模拟和初步实验结果,我们相信所提出的方法可以很容易地应用于任何高度多态的基因系统。