Kim Jihee, Ahn Seungkirl, Rajagopal Keshava, Lefkowitz Robert J
Department of Medicine, Duke University Medical Center, Durham, NC 27710, USA.
J Biol Chem. 2009 May 1;284(18):11953-62. doi: 10.1074/jbc.M808176200. Epub 2009 Mar 2.
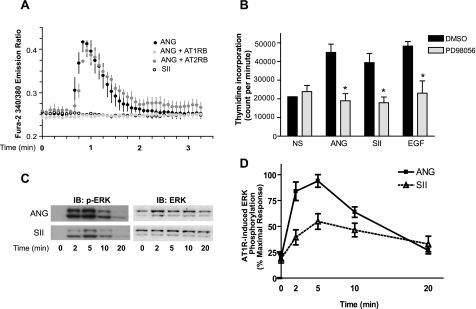
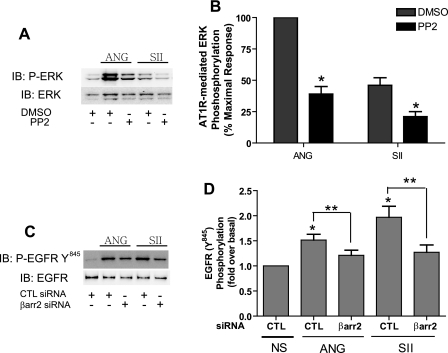
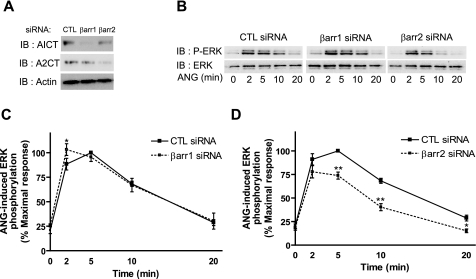
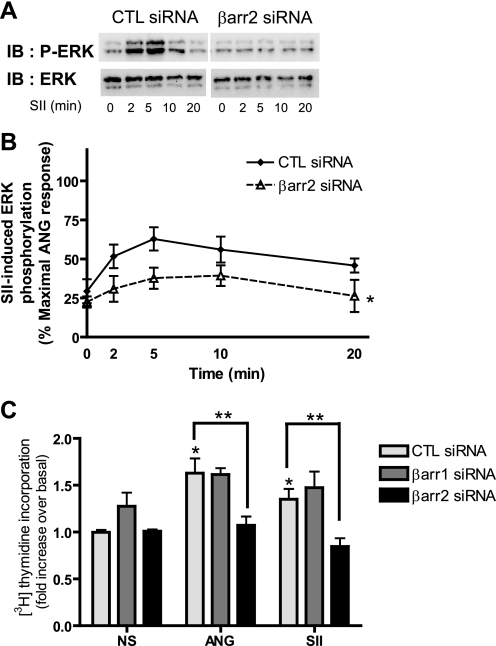
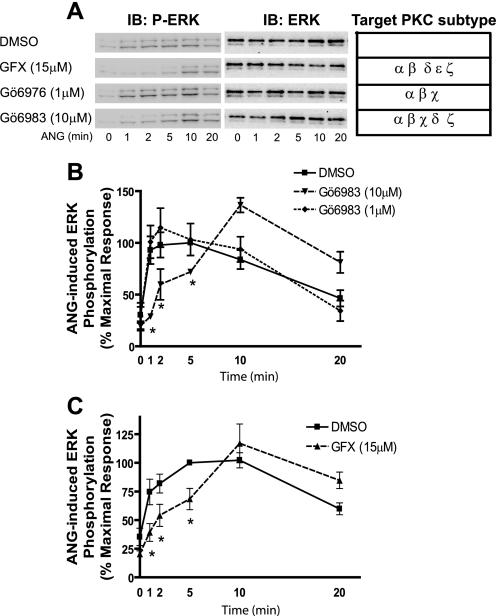
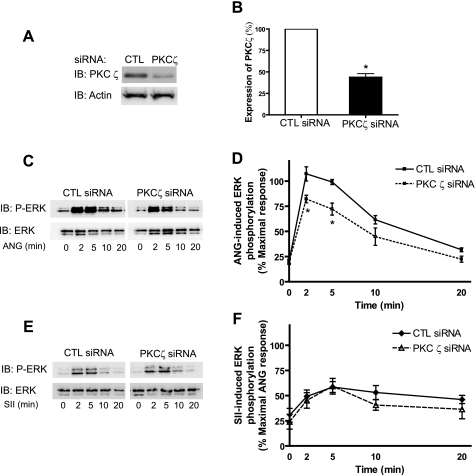
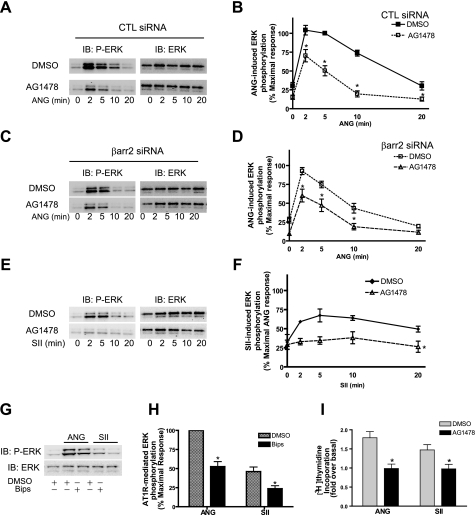
Recent studies in receptor-transfected cell lines have demonstrated that extracellular signal-regulated kinase (ERK) activation by angiotensin type 1A receptor and other G protein-coupled receptors can be mediated by both G protein-dependent and beta-arrestin-dependent mechanisms. However, few studies have explored these mechanisms in primary cultured cells expressing endogenous levels of receptors. Accordingly, here we utilized the beta-arrestin biased agonist for the angiotensin type 1A receptor, SII-angiotensin (SII), and RNA interference techniques to investigate angiotensin II (ANG)-activated beta-arrestin-mediated mitogenic signaling pathways in rat vascular smooth muscle cells. Both ANG and SII induced DNA synthesis via the ERK activation cascade. Even though SII cannot induce calcium influx (G protein activation) after receptor stimulation, it does cause ERK activation, although less robustly than ANG. Activation by both ligands is diminished by depletion of beta-arrestin2 by small interfering RNA, although the effect is more complete with SII. ERK activation at early time points but not later time points is strongly inhibited by those protein kinase C inhibitors that can block protein kinase Czeta. Moreover, ANG- and SII-mediated ERK activation require transactivation of the epidermal growth factor receptor via metalloprotease 2/9 and Src kinase. beta-Arrestin2 facilitates ANG and SII stimulation of Src-mediated phosphorylation of Tyr-845 on the EGFR, a known site for Src phosphorylation. These studies delineate a convergent mechanism by which G protein-dependent and beta-arrestin-dependent pathways can independently mediate ERK-dependent transactivation of the EGFR in vascular smooth muscle cells thus controlling cellular proliferative responses.
近期在受体转染细胞系中的研究表明,1A型血管紧张素受体及其他G蛋白偶联受体激活细胞外信号调节激酶(ERK)可通过G蛋白依赖性和β-抑制蛋白依赖性两种机制介导。然而,很少有研究在表达内源性受体水平的原代培养细胞中探究这些机制。因此,我们利用1A型血管紧张素受体的β-抑制蛋白偏向性激动剂SII-血管紧张素(SII)以及RNA干扰技术,来研究血管紧张素II(ANG)激活的β-抑制蛋白介导的大鼠血管平滑肌细胞有丝分裂信号通路。ANG和SII均通过ERK激活级联反应诱导DNA合成。尽管受体刺激后SII不能诱导钙内流(G蛋白激活),但它确实能引起ERK激活,尽管其激活程度不如ANG强烈。小干扰RNA使β-抑制蛋白2耗竭后,两种配体的激活作用均减弱,不过SII的这种作用更完全。能阻断蛋白激酶Cζ的蛋白激酶C抑制剂可强烈抑制早期而非后期的ERK激活。此外,ANG和SII介导的ERK激活需要通过金属蛋白酶2/9和Src激酶对表皮生长因子受体进行反式激活。β-抑制蛋白2促进ANG和SII刺激Src介导的表皮生长因子受体上Tyr-845位点的磷酸化,该位点是已知的Src磷酸化位点。这些研究描绘了一种趋同机制,通过该机制,G蛋白依赖性和β-抑制蛋白依赖性途径可独立介导血管平滑肌细胞中表皮生长因子受体的ERK依赖性反式激活,从而控制细胞增殖反应。