Weatherly D Brent, Boehlke Courtney, Tarleton Rick L
Center for Tropical and Emerging Global Diseases, University of Georgia, Athens, GA, USA.
BMC Genomics. 2009 Jun 1;10:255. doi: 10.1186/1471-2164-10-255.
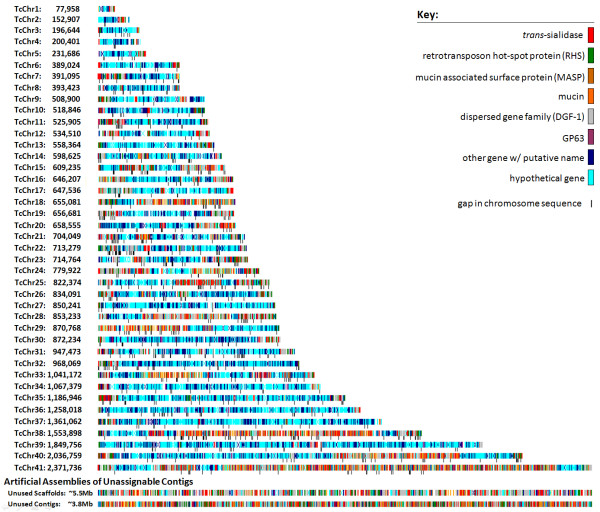
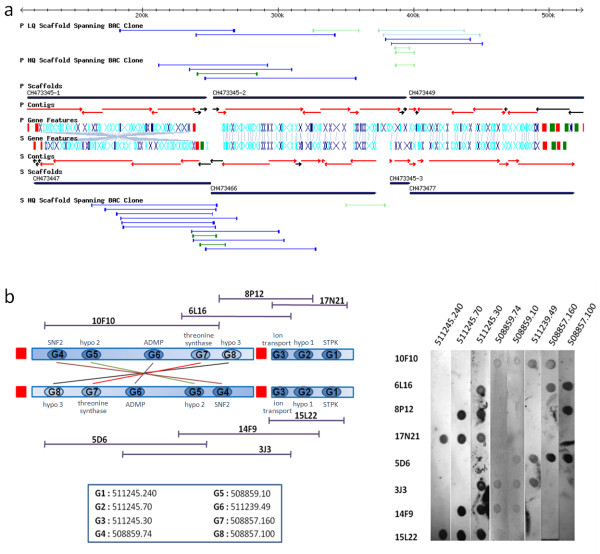
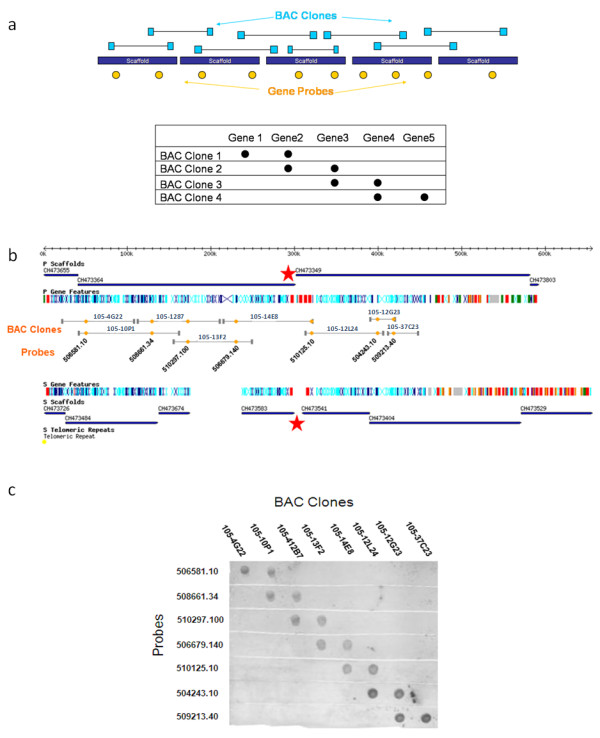
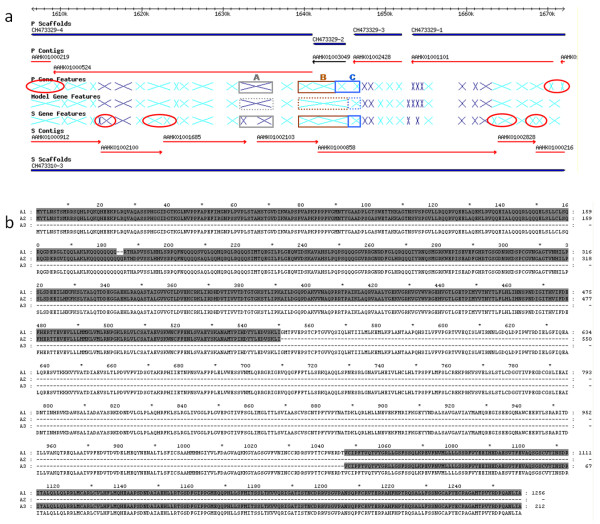
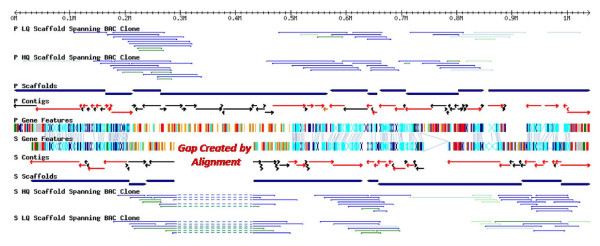
In contrast to the essentially fully assembled genome sequences of the kinetoplastid pathogens Leishmania major and Trypanosoma brucei the assembly of the Trypanosoma cruzi genome has been hindered by its repetitive nature and the fact that the reference strain (CL Brener) is a hybrid of two distinct lineages. In this work, the majority of the contigs and scaffolds were assembled into pairs of homologous chromosomes based on predicted parental haplotype, inference from TriTryp synteny maps and the use of end sequences from T. cruzi BAC libraries.
Ultimately, 41 pairs of chromosomes were assembled using this approach, a number in agreement with the predicted number of T. cruzi chromosomes based upon pulse field gel analysis, with over 90% (21133 of 23216) of the genes annotated in the genome represented. The approach was substantiated through the use of Southern blot analysis to confirm the mapping of BAC clones using as probes the genes they are predicted to contain, and each chromosome construction was visually validated to ensure sufficient evidence was present to support the organization. While many members of large gene families are incorporated into the chromosome assemblies, the majority of genes excluded from the chromosomes belong to gene families, as these genes are frequently impossible to accurately position.
Now assembled, these chromosomes bring T. cruzi to the same level of organization as its kinetoplastid relatives and have been used as the basis for the T. cruzi genome in TriTrypDB, a trypanosome database of EuPathDB. In addition, they will provide the foundation for analyses such as reverse genetics, where the location of genes and their alleles and/or paralogues is necessary and comparative genome hybridization analyses (CGH), where a chromosome-level view of the genome is ideal.
与动基体病原体硕大利什曼原虫和布氏锥虫基本完全组装好的基因组序列不同,克氏锥虫基因组的组装因其重复性质以及参考菌株(CL Brener)是两个不同谱系的杂交体这一事实而受到阻碍。在这项研究中,基于预测的亲本单倍型、从三锥虫同线图推断以及使用克氏锥虫细菌人工染色体(BAC)文库的末端序列,大多数重叠群和支架被组装成同源染色体对。
最终,使用这种方法组装了41对染色体,这一数量与基于脉冲场凝胶分析预测的克氏锥虫染色体数量一致,基因组中注释的基因有超过90%(23216个中的21133个)得到了体现。通过使用Southern印迹分析来确认BAC克隆的定位,以它们预测包含的基因作为探针,从而证实了该方法,并且对每个染色体构建进行了可视化验证,以确保有足够的证据支持其组织形式。虽然许多大基因家族的成员被纳入了染色体组装中,但大多数被排除在染色体之外的基因属于基因家族,因为这些基因往往无法准确定位。
现在这些染色体已组装完成将克氏锥虫提升到了与其动基体亲属相同的组织水平,并已被用作锥虫数据库EuPathDB中克氏锥虫基因组的基础。此外,它们将为诸如反向遗传学(其中基因及其等位基因和/或旁系同源物的位置是必要的)和比较基因组杂交分析(CGH,其中基因组的染色体水平视图理想)等分析提供基础。