Homer Nils, Merriman Barry, Nelson Stanley F
Department of Computer Science, University of California Los Angeles, Los Angeles, California 90095, USA.
BMC Bioinformatics. 2009 Jun 9;10:175. doi: 10.1186/1471-2105-10-175.
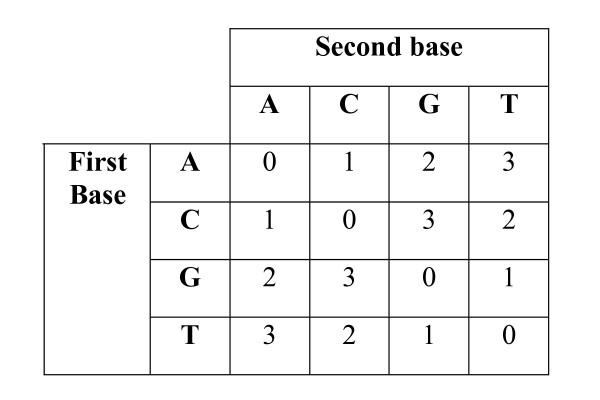
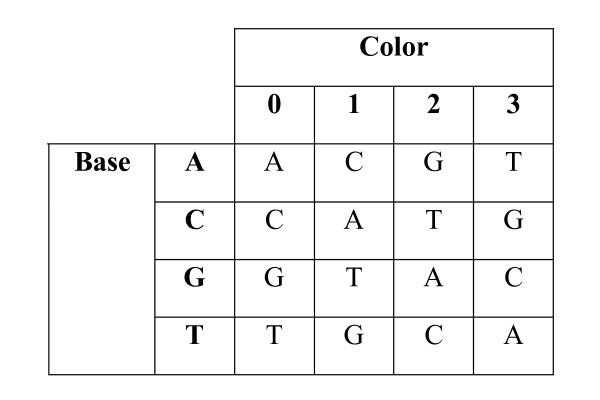
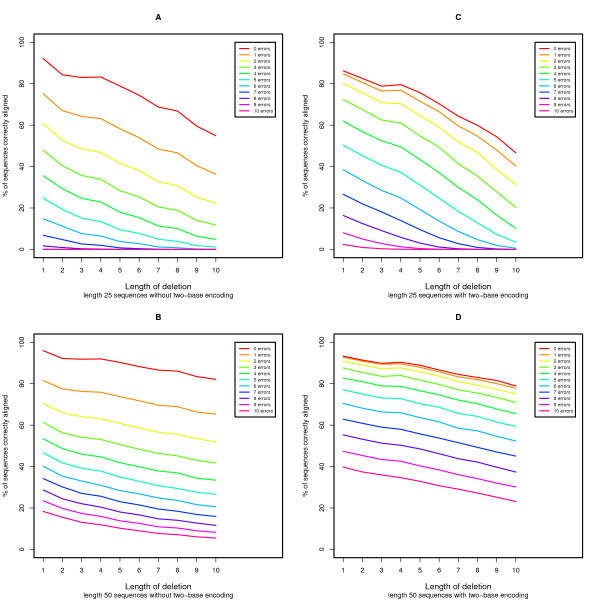
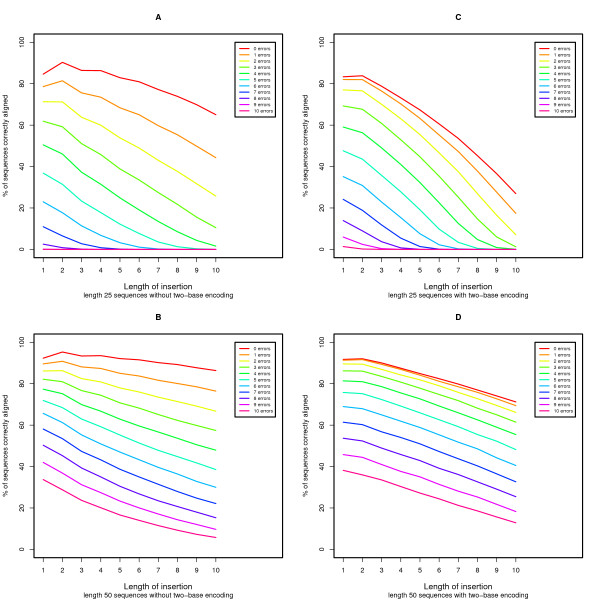
DNA sequence comparison is based on optimal local alignment of two sequences using a similarity score. However, some new DNA sequencing technologies do not directly measure the base sequence, but rather an encoded form, such as the two-base encoding considered here. In order to compare such data to a reference sequence, the data must be decoded into sequence. The decoding is deterministic, but the possibility of measurement errors requires searching among all possible error modes and resulting alignments to achieve an optimal balance of fewer errors versus greater sequence similarity.
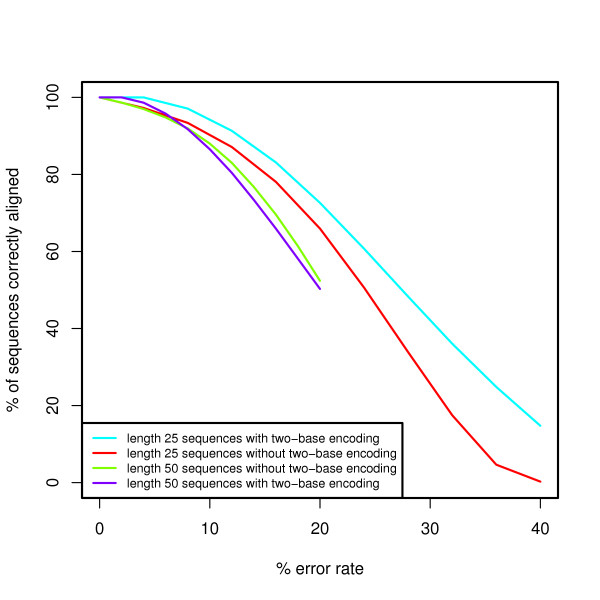
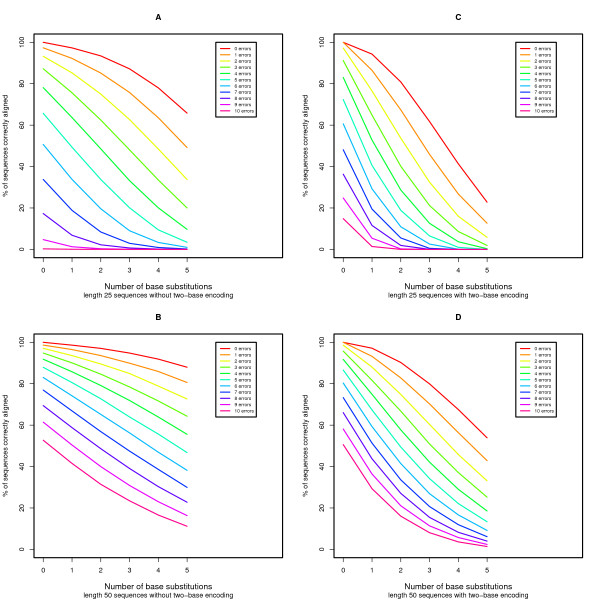
We present an extension of the standard dynamic programming method for local alignment, which simultaneously decodes the data and performs the alignment, maximizing a similarity score based on a weighted combination of errors and edits, and allowing an affine gap penalty. We also present simulations that demonstrate the performance characteristics of our two base encoded alignment method and contrast those with standard DNA sequence alignment under the same conditions.
The new local alignment algorithm for two-base encoded data has substantial power to properly detect and correct measurement errors while identifying underlying sequence variants, and facilitating genome re-sequencing efforts based on this form of sequence data.
DNA序列比较基于使用相似性得分对两个序列进行最优局部比对。然而,一些新的DNA测序技术并不直接测量碱基序列,而是测量一种编码形式,比如这里所考虑的双碱基编码。为了将此类数据与参考序列进行比较,必须将数据解码为序列。解码是确定性的,但测量误差的可能性要求在所有可能的错误模式及由此产生的比对中进行搜索,以在较少错误与较高序列相似性之间实现最优平衡。
我们提出了一种对局部比对标准动态规划方法的扩展,该方法同时对数据进行解码并执行比对,基于错误和编辑的加权组合最大化相似性得分,并允许仿射空位罚分。我们还展示了模拟结果,这些结果证明了我们的双碱基编码比对方法的性能特征,并在相同条件下将其与标准DNA序列比对进行了对比。
针对双碱基编码数据的新局部比对算法在识别潜在序列变异的同时,具有强大的能力来正确检测和校正测量误差,并有助于基于这种序列数据形式的基因组重测序工作。