Division of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA.
BMC Bioinformatics. 2012 Nov 9;13:293. doi: 10.1186/1471-2105-13-293.
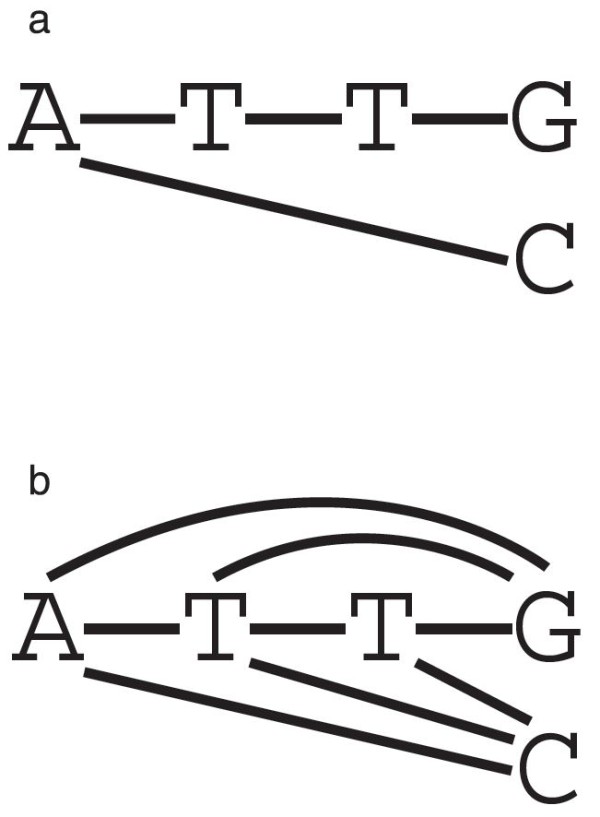
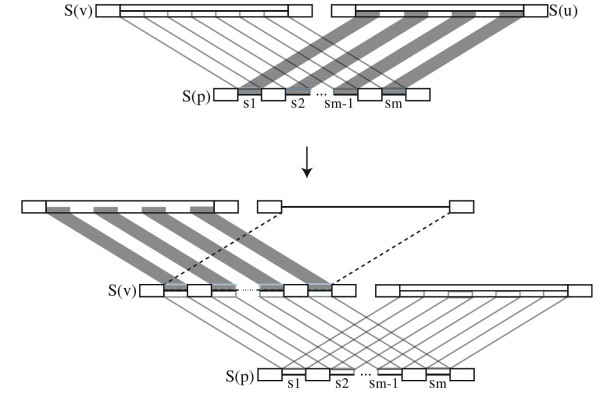
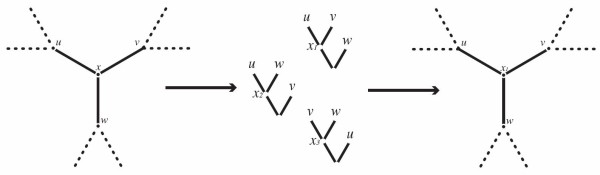
The inference of homologies among DNA sequences, that is, positions in multiple genomes that share a common evolutionary origin, is a crucial, yet difficult task facing biologists. Its computational counterpart is known as the multiple sequence alignment problem. There are various criteria and methods available to perform multiple sequence alignments, and among these, the minimization of the overall cost of the alignment on a phylogenetic tree is known in combinatorial optimization as the Tree Alignment Problem. This problem typically occurs as a subproblem of the Generalized Tree Alignment Problem, which looks for the tree with the lowest alignment cost among all possible trees. This is equivalent to the Maximum Parsimony problem when the input sequences are not aligned, that is, when phylogeny and alignments are simultaneously inferred.
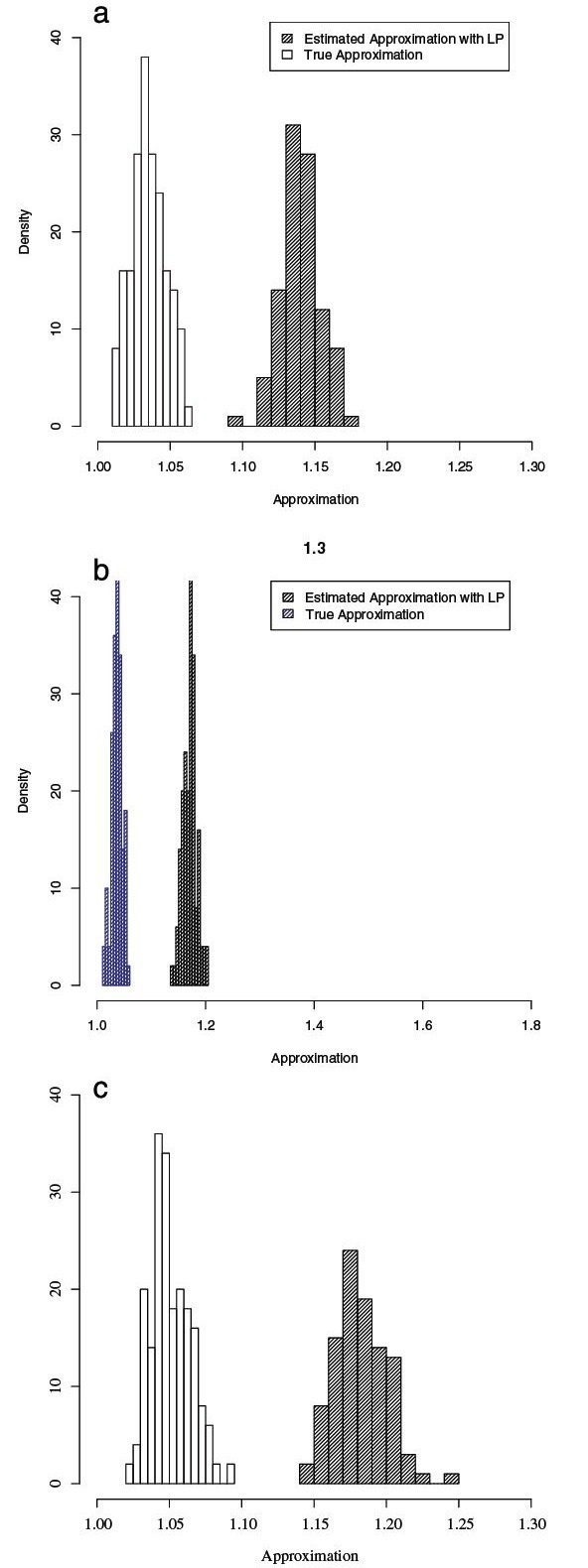
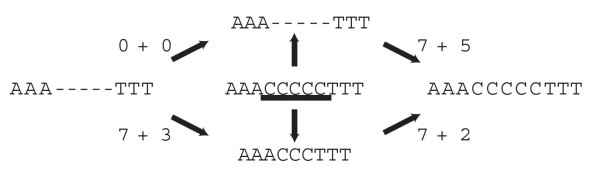
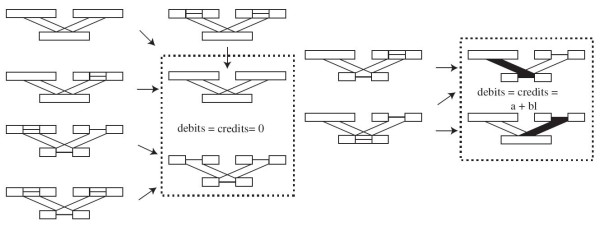
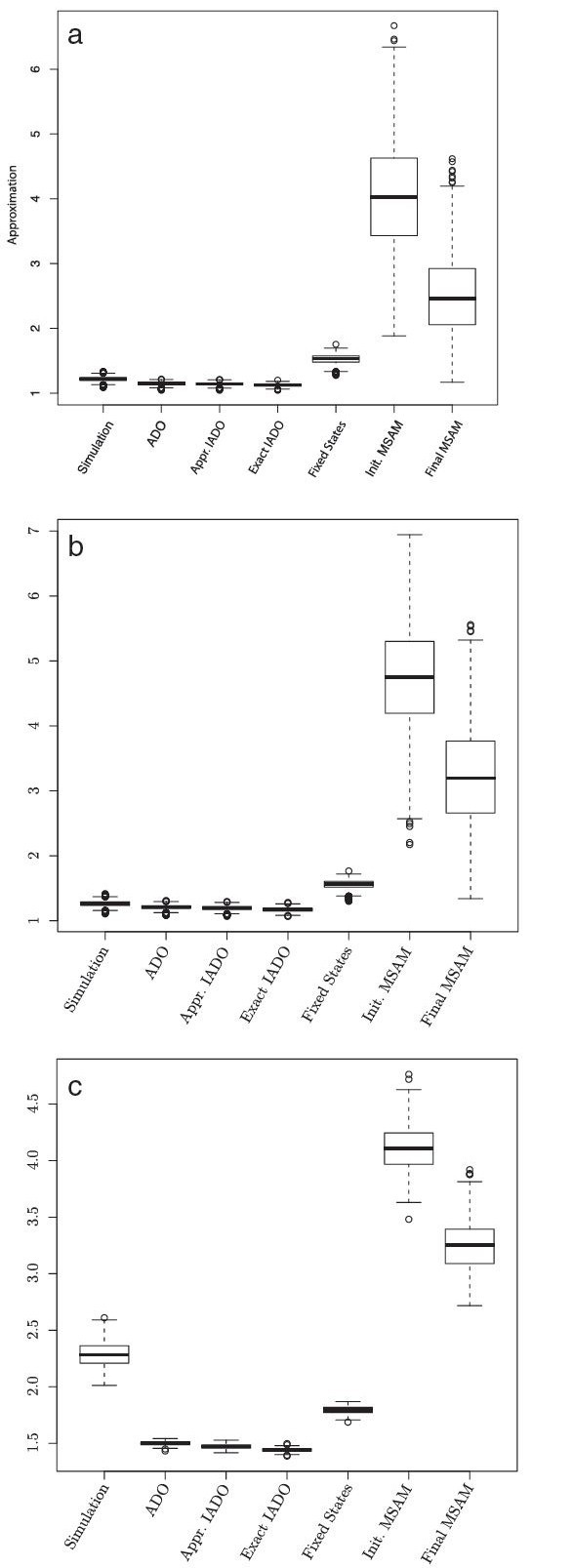
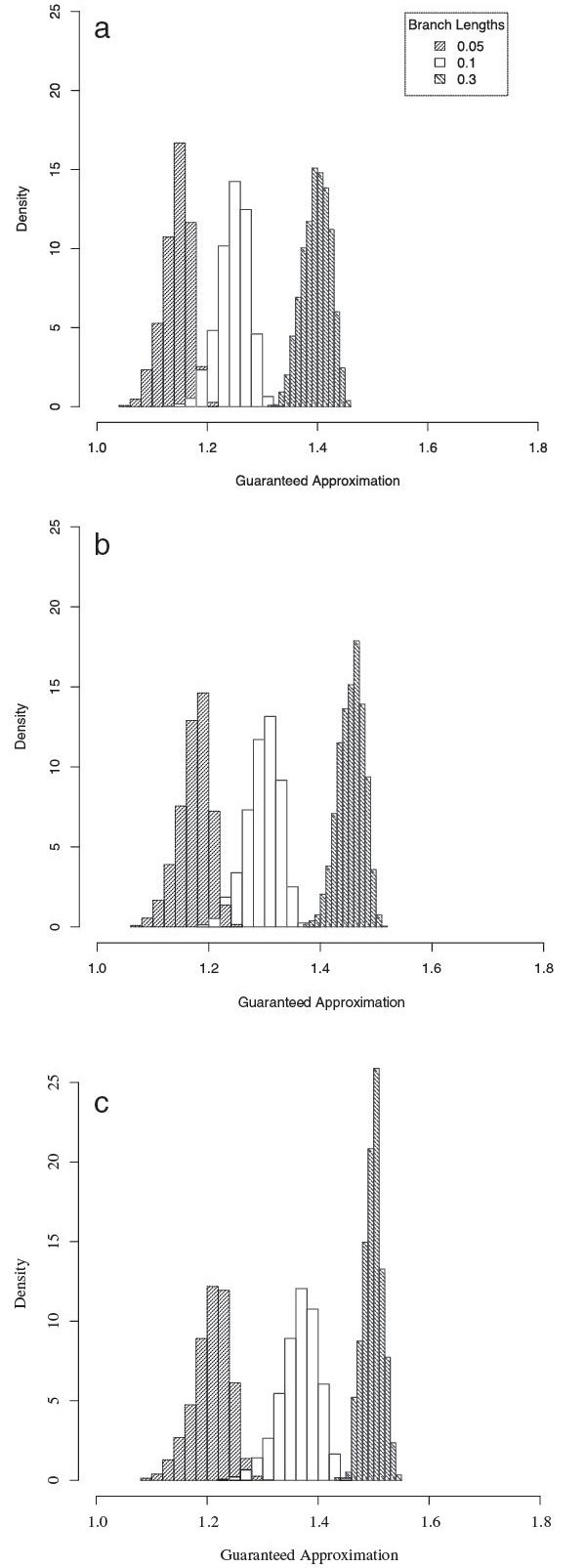
For large data sets, a popular heuristic is Direct Optimization (DO). DO provides a good tradeoff between speed, scalability, and competitive scores, and is implemented in the computer program POY. All other (competitive) algorithms have greater time complexities compared to DO. Here, we introduce and present experiments a new algorithm Affine-DO to accommodate the indel (alignment gap) models commonly used in phylogenetic analysis of molecular sequence data. Affine-DO has the same time complexity as DO, but is correctly suited for the affine gap edit distance. We demonstrate its performance with more than 330,000 experimental tests. These experiments show that the solutions of Affine-DO are close to the lower bound inferred from a linear programming solution. Moreover, iterating over a solution produced using Affine-DO shows little improvement.
Our results show that Affine-DO is likely producing near-optimal solutions, with approximations within 10% for sequences with small divergence, and within 30% for random sequences, for which Affine-DO produced the worst solutions. The Affine-DO algorithm has the necessary scalability and optimality to be a significant improvement in the real-world phylogenetic analysis of sequence data.
推断 DNA 序列之间的同源性,即多个基因组中共享共同进化起源的位置,是生物学家面临的一项关键但困难的任务。其计算对应物被称为多序列比对问题。有各种标准和方法可用于进行多序列比对,其中,在系统发育树上最小化比对的总代价被称为树比对问题。在组合优化中,这个问题通常作为广义树比对问题的子问题出现,该问题在所有可能的树中寻找具有最低比对代价的树。当输入序列未对齐时,即同时推断系统发育和比对时,这相当于最大简约问题。
对于大型数据集,一种流行的启发式方法是直接优化(DO)。DO 在速度、可扩展性和竞争分数之间提供了良好的折衷,并且在计算机程序 POY 中实现。与 DO 相比,所有其他(竞争)算法的时间复杂度都更高。在这里,我们引入并展示了一种新算法——仿射-DO,以适应分子序列数据系统发育分析中常用的插入缺失(比对间隙)模型。仿射-DO 具有与 DO 相同的时间复杂度,但非常适合仿射间隙编辑距离。我们通过超过 330000 次实验测试展示了它的性能。这些实验表明,仿射-DO 的解决方案接近从线性规划解决方案推断出的下限。此外,在使用仿射-DO 生成的解决方案上迭代几乎没有改进。
我们的结果表明,仿射-DO 很可能产生接近最优的解决方案,对于具有小分歧的序列,其近似值在 10%以内,对于随机序列,其近似值在 30%以内,对于仿射-DO 生成的最差解决方案,其近似值在 30%以内。仿射-DO 算法具有必要的可扩展性和最优性,可以显著改进序列数据的实际系统发育分析。