Bayden Alexander S, Fornabaio Micaela, Scarsdale J Neel, Kellogg Glen E
Department of Medicinal Chemistry & Institute for Structural Biology and Drug Discovery, Virginia Commonwealth University, Richmond, VA 23298-0540, USA.
J Comput Aided Mol Des. 2009 Sep;23(9):621-32. doi: 10.1007/s10822-009-9270-7. Epub 2009 Jun 25.
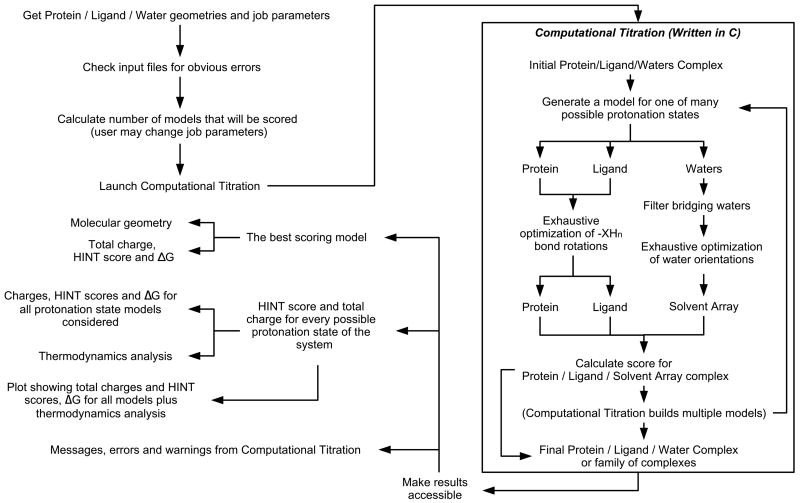
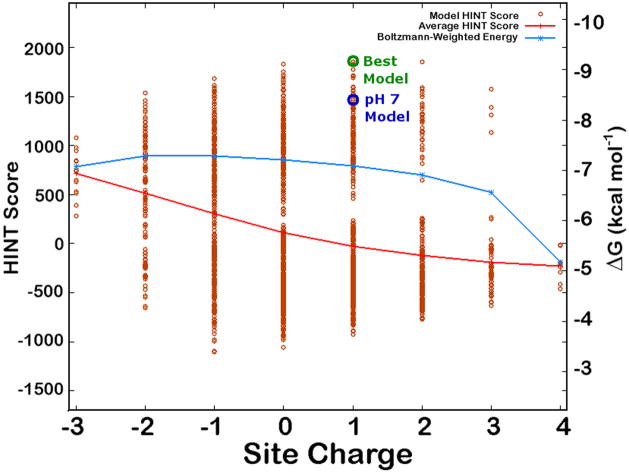
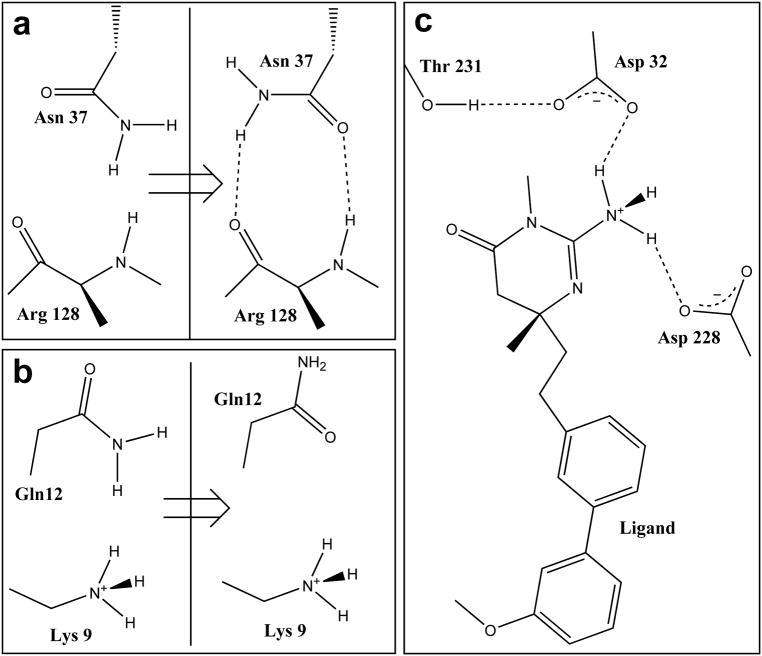
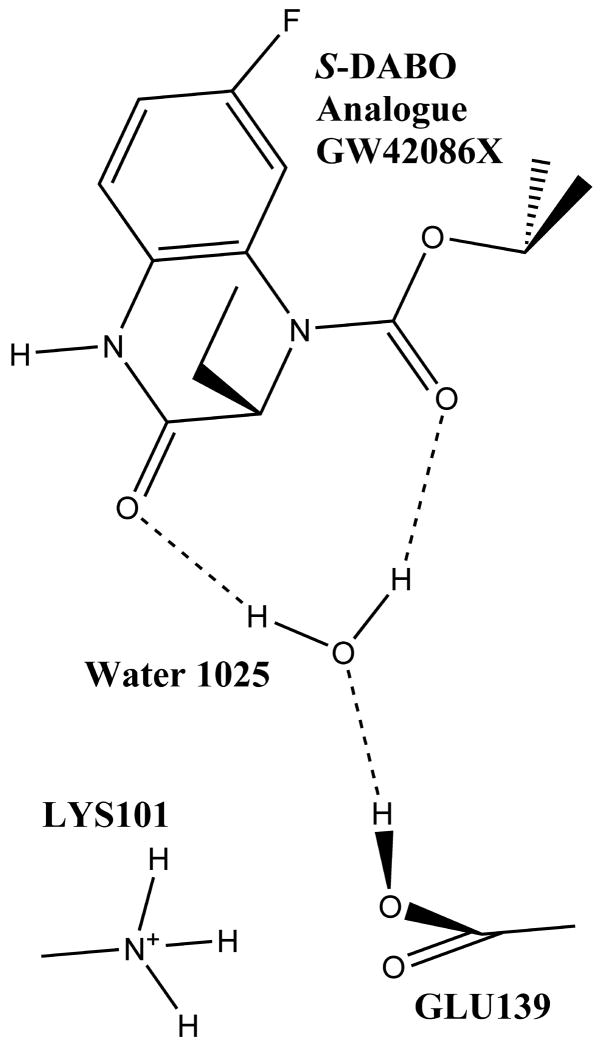
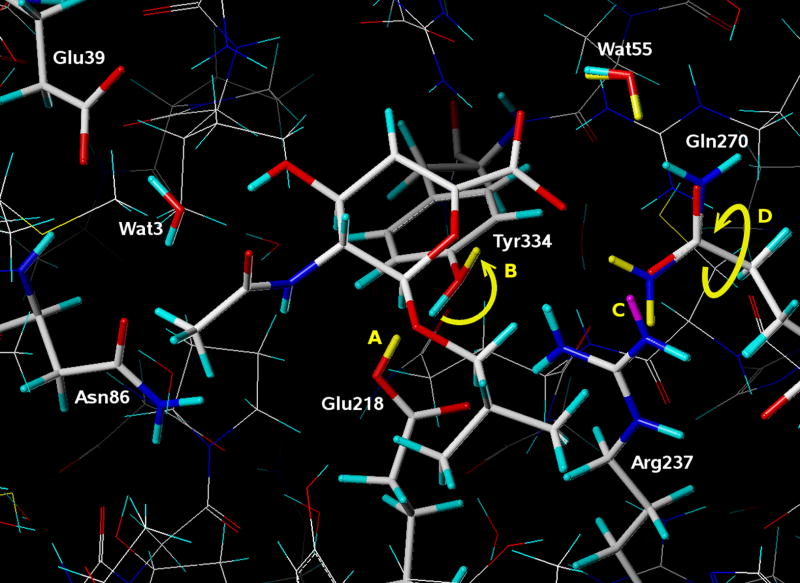
A public web server performing computational titration at the active site in a protein-ligand complex has been implemented. This calculation is based on the Hydropathic interaction noncovalent force field. From 3D coordinate data for the protein, ligand and bridging waters (if available), the server predicts the best combination of protonation states for each ionizable residue and/or ligand functional group as well as the Gibbs free energy of binding for the ionization-optimized protein-ligand complex. The 3D structure for the modified molecules is available as output. In addition, a graph depicting how this energy changes with acidity, i.e., as a function of added protons, can be obtained. This data may prove to be of use in preparing models for virtual screening and molecular docking. A few illustrative examples are presented. In beta secretase (2va7) computational titration flipped the amide groups of Gln12 and Asn37 and protonated a ligand amine yielding an improvement of 6.37 kcal mol(-1) in the protein-ligand binding score. Protonation of Glu139 in mutant HIV-1 reverse transcriptase (2opq) allows a water bridge between the protein and inhibitor that increases the protein-ligand interaction score by 0.16 kcal mol(-1). In human sialidase NEU2 complexed with an isobutyl ether mimetic inhibitor (2f11) computational titration suggested that protonating Glu218, deprotonating Arg237, flipping the amide bond on Tyr334, and optimizing the positions of several other polar protons would increase the protein-ligand interaction score by 0.71 kcal mol(-1).
已经实现了一个在蛋白质 - 配体复合物的活性位点进行计算滴定的公共网络服务器。该计算基于疏水相互作用非共价力场。根据蛋白质、配体和桥连水(如果有的话)的三维坐标数据,服务器预测每个可电离残基和/或配体官能团的最佳质子化状态组合,以及电离优化后的蛋白质 - 配体复合物的结合吉布斯自由能。修饰分子的三维结构可作为输出提供。此外,还可以获得一个描绘该能量如何随酸度变化(即作为添加质子的函数)的图表。这些数据可能在准备虚拟筛选和分子对接模型方面有用。给出了一些说明性示例。在β-分泌酶(2va7)中,计算滴定使Gln12和Asn37的酰胺基团翻转,并使一个配体胺质子化,从而使蛋白质 - 配体结合分数提高了6.37千卡/摩尔(-1)。突变型HIV - 1逆转录酶(2opq)中Glu139的质子化允许在蛋白质和抑制剂之间形成水桥,使蛋白质 - 配体相互作用分数增加了0.16千卡/摩尔(-1)。在与异丁基醚模拟抑制剂复合的人唾液酸酶NEU2(2f11)中,计算滴定表明,使Glu218质子化、使Arg237去质子化、翻转Tyr334上的酰胺键以及优化其他几个极性质子的位置将使蛋白质 - 配体相互作用分数提高0.71千卡/摩尔(-1)。