Kim Jong-Il, Ju Young Seok, Park Hansoo, Kim Sheehyun, Lee Seonwook, Yi Jae-Hyuk, Mudge Joann, Miller Neil A, Hong Dongwan, Bell Callum J, Kim Hye-Sun, Chung In-Soon, Lee Woo-Chung, Lee Ji-Sun, Seo Seung-Hyun, Yun Ji-Young, Woo Hyun Nyun, Lee Heewook, Suh Dongwhan, Lee Seungbok, Kim Hyun-Jin, Yavartanoo Maryam, Kwak Minhye, Zheng Ying, Lee Mi Kyeong, Park Hyunjun, Kim Jeong Yeon, Gokcumen Omer, Mills Ryan E, Zaranek Alexander Wait, Thakuria Joseph, Wu Xiaodi, Kim Ryan W, Huntley Jim J, Luo Shujun, Schroth Gary P, Wu Thomas D, Kim HyeRan, Yang Kap-Seok, Park Woong-Yang, Kim Hyungtae, Church George M, Lee Charles, Kingsmore Stephen F, Seo Jeong-Sun
Genomic Medicine Institute, Medical Research Center, Seoul National University, Seoul 110-799, Korea.
Nature. 2009 Aug 20;460(7258):1011-5. doi: 10.1038/nature08211. Epub 2009 Jul 8.
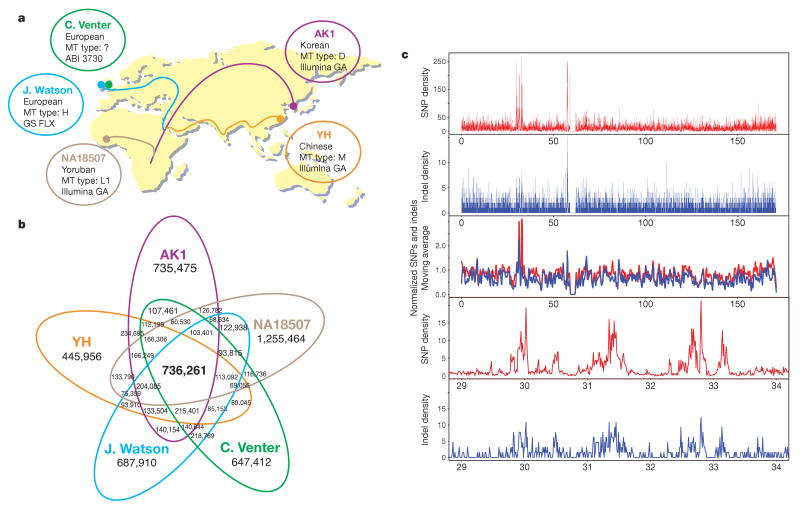
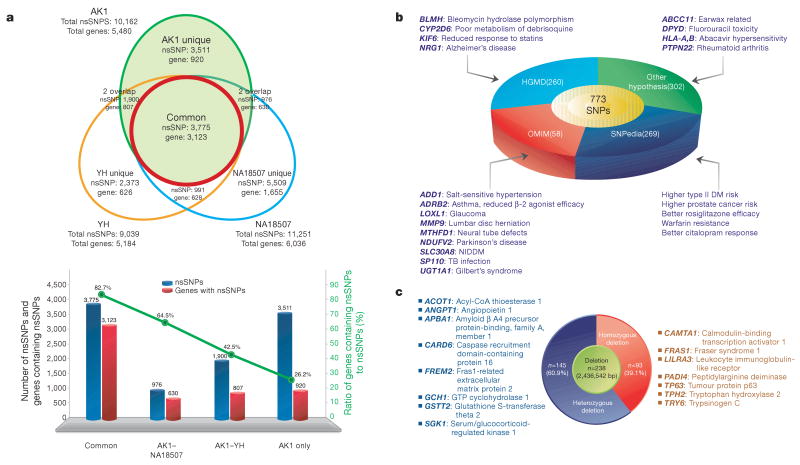
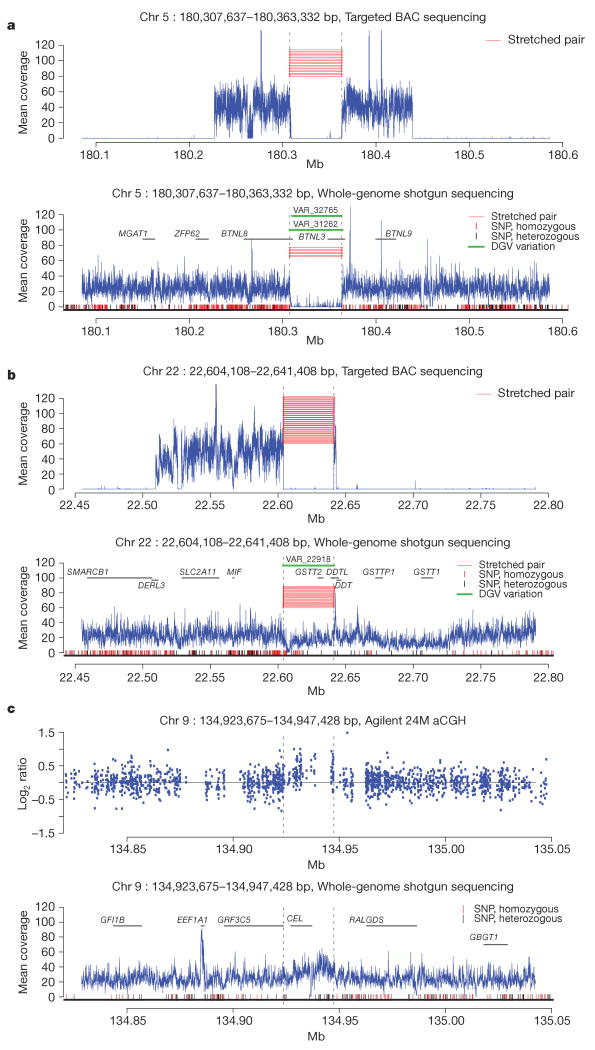
Recent advances in sequencing technologies have initiated an era of personal genome sequences. To date, human genome sequences have been reported for individuals with ancestry in three distinct geographical regions: a Yoruba African, two individuals of northwest European origin, and a person from China. Here we provide a highly annotated, whole-genome sequence for a Korean individual, known as AK1. The genome of AK1 was determined by an exacting, combined approach that included whole-genome shotgun sequencing (27.8x coverage), targeted bacterial artificial chromosome sequencing, and high-resolution comparative genomic hybridization using custom microarrays featuring more than 24 million probes. Alignment to the NCBI reference, a composite of several ethnic clades, disclosed nearly 3.45 million single nucleotide polymorphisms (SNPs), including 10,162 non-synonymous SNPs, and 170,202 deletion or insertion polymorphisms (indels). SNP and indel densities were strongly correlated genome-wide. Applying very conservative criteria yielded highly reliable copy number variants for clinical considerations. Potential medical phenotypes were annotated for non-synonymous SNPs, coding domain indels, and structural variants. The integration of several human whole-genome sequences derived from several ethnic groups will assist in understanding genetic ancestry, migration patterns and population bottlenecks.
测序技术的最新进展开启了个人基因组序列的时代。迄今为止,已报道了来自三个不同地理区域的个体的人类基因组序列:一名约鲁巴非洲人、两名西北欧裔个体以及一名中国人。在此,我们提供了一份针对一名韩国个体(称为AK1)的高度注释的全基因组序列。AK1的基因组通过一种严格的组合方法确定,该方法包括全基因组鸟枪法测序(27.8倍覆盖)、靶向细菌人工染色体测序以及使用具有超过2400万个探针的定制微阵列进行高分辨率比较基因组杂交。与NCBI参考序列(几个种族分支的复合体)比对,发现了近345万个单核苷酸多态性(SNP),包括10162个非同义SNP以及170202个缺失或插入多态性(indel)。SNP和indel密度在全基因组范围内高度相关。应用非常保守的标准产生了用于临床考虑的高度可靠的拷贝数变异。对非同义SNP、编码域indel和结构变异进行了潜在医学表型注释。整合来自多个种族群体的几个人类全基因组序列将有助于理解遗传谱系、迁移模式和种群瓶颈。