Zhang Jing, Liu Bing, Jiang Xingpeng, Zhao Huizhi, Fan Ming, Fan Zhenjie, Lee J Jack, Jiang Tao, Jiang Tianzi, Song Sonya Wei
LIAMA Center for Computational Medicine, National Laboratory of Pattern Recognition, Institute of Automation, Chinese Academy of Sciences, Beijing, the People's Republic of China.
PLoS One. 2009 Jul 17;4(7):e6274. doi: 10.1371/journal.pone.0006274.
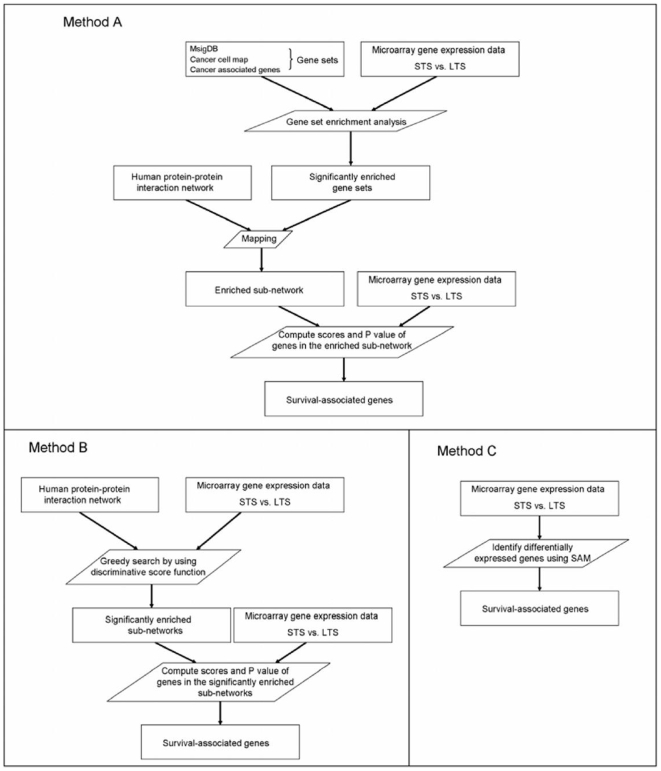
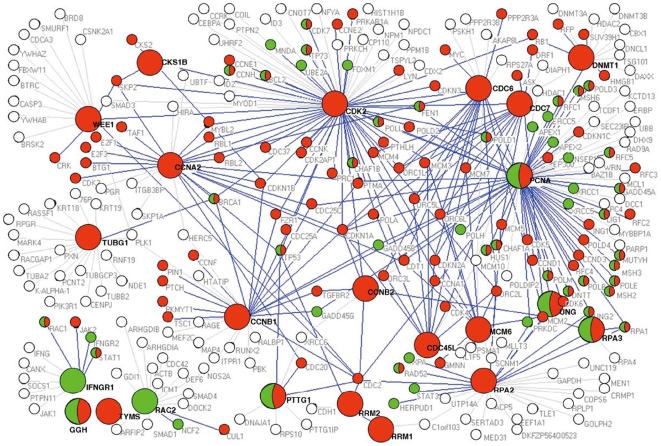
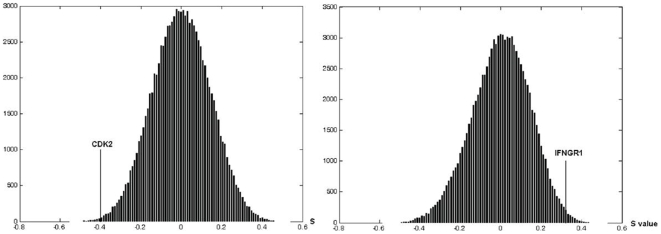
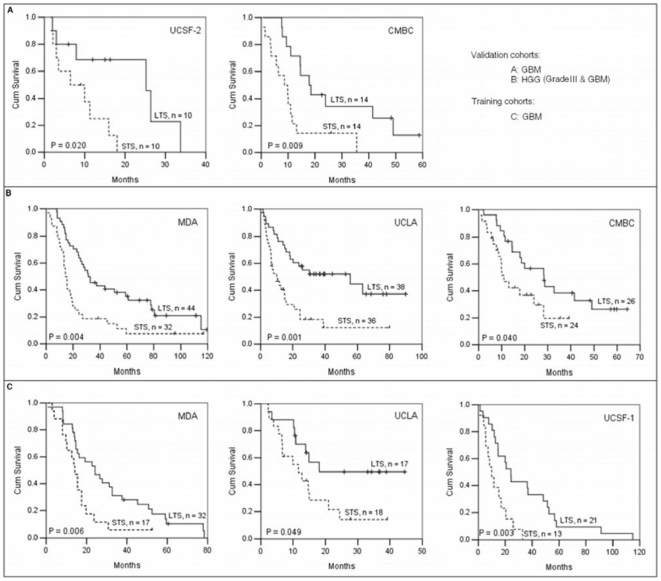
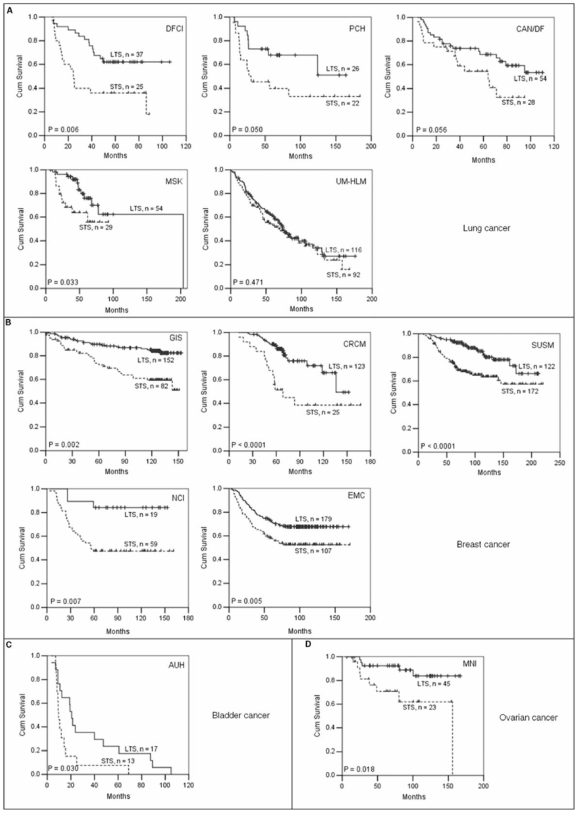
Accurate prediction of survival of cancer patients is still a key open problem in clinical research. Recently, many large-scale gene expression clusterings have identified sets of genes reportedly predictive of prognosis; however, those gene sets shared few genes in common and were poorly validated using independent data. We have developed a systems biology-based approach by using either combined gene sets and the protein interaction network (Method A) or the protein network alone (Method B) to identify common prognostic genes based on microarray gene expression data of glioblastoma multiforme and compared with differential gene expression clustering (Method C). Validations of prediction performance show that the 23-prognostic gene classifier identified by Method A outperforms other gene classifiers identified by Methods B and C or previously reported for gliomas on 17 of 20 independent sample cohorts across five tumor types. We also find that among the 23 genes are 21 related to cellular proliferation and two related to response to stress/immune response. We further find that the increased expression of the 21 genes and the decreased expression of the other two genes are associated with poorer survival, which is supportive with the notion that cellular proliferation and immune response contribute to a significant portion of predictive power of prognostic classifiers. Our results demonstrate that the systems biology-based approach enables to identify common survival-associated genes.
准确预测癌症患者的生存率仍是临床研究中一个关键的开放性问题。最近,许多大规模基因表达聚类研究已鉴定出据报道可预测预后的基因集;然而,这些基因集之间几乎没有共同的基因,并且使用独立数据进行的验证效果不佳。我们开发了一种基于系统生物学的方法,通过使用组合基因集和蛋白质相互作用网络(方法A)或仅使用蛋白质网络(方法B),基于多形性胶质母细胞瘤的微阵列基因表达数据来识别常见的预后基因,并与差异基因表达聚类(方法C)进行比较。预测性能的验证表明,通过方法A鉴定出的23个预后基因分类器在五种肿瘤类型的20个独立样本队列中的17个队列中,优于通过方法B和C鉴定出的其他基因分类器或先前报道的胶质瘤基因分类器。我们还发现,在这23个基因中,有21个与细胞增殖相关,2个与应激反应/免疫反应相关。我们进一步发现,这21个基因的表达增加和另外两个基因的表达降低与较差的生存率相关,这支持了细胞增殖和免疫反应在预后分类器的预测能力中占很大比例的观点。我们的结果表明,基于系统生物学的方法能够识别常见的生存相关基因。