Biozentrum, University of Basel, and Swiss Institute of Bioinformatics, Klingelbergstrasse 50/70, 4056-CH, Basel, Switzerland.
Genome Biol. 2009;10(7):R79. doi: 10.1186/gb-2009-10-7-r79. Epub 2009 Jul 22.
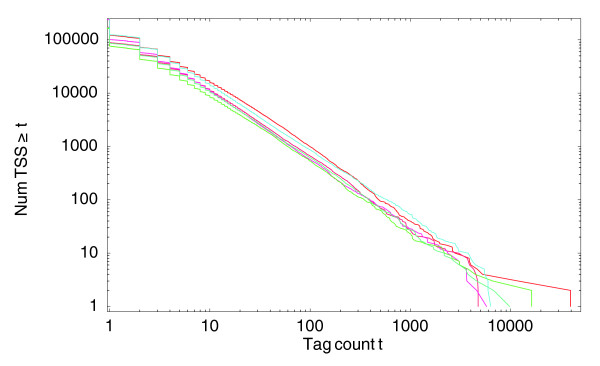
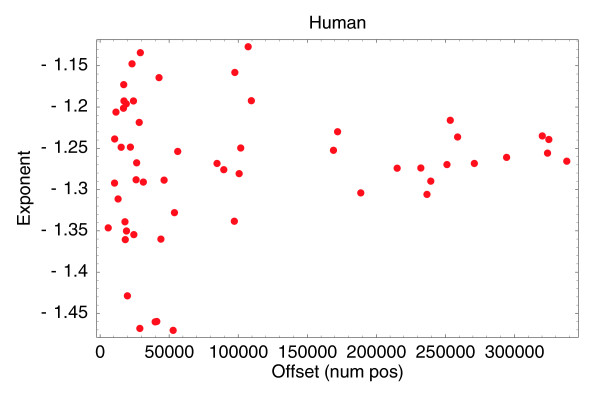
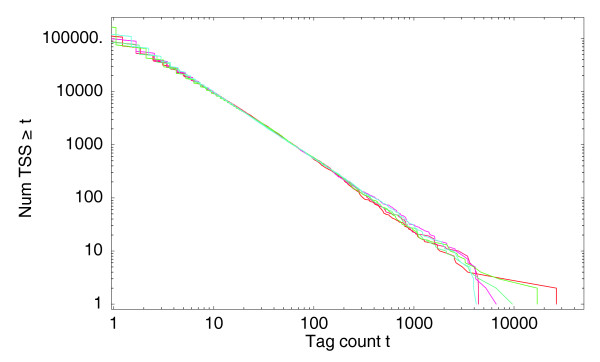
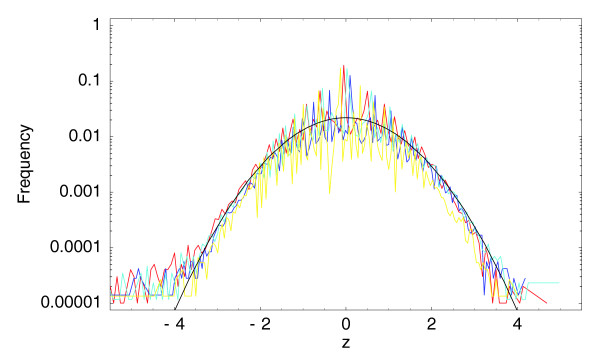
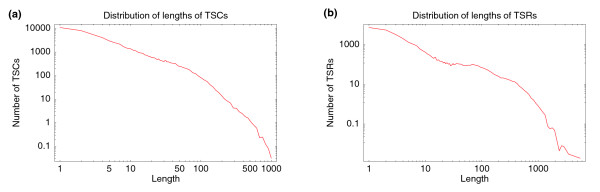
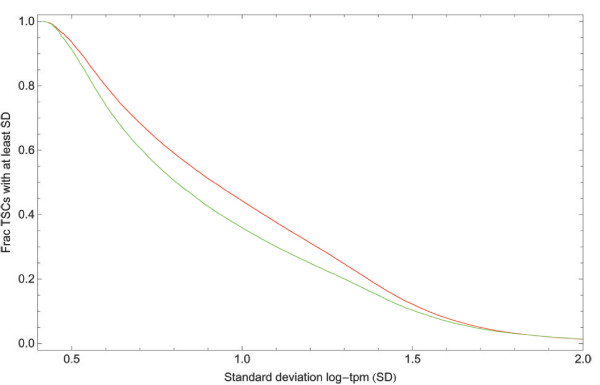
With the advent of ultra high-throughput sequencing technologies, increasingly researchers are turning to deep sequencing for gene expression studies. Here we present a set of rigorous methods for normalization, quantification of noise, and co-expression analysis of deep sequencing data. Using these methods on 122 cap analysis of gene expression (CAGE) samples of transcription start sites, we construct genome-wide 'promoteromes' in human and mouse consisting of a three-tiered hierarchy of transcription start sites, transcription start clusters, and transcription start regions.
随着超高通量测序技术的出现,越来越多的研究人员开始将深度测序应用于基因表达研究。在这里,我们提出了一套严格的方法,用于对深度测序数据进行归一化、噪声量化和共表达分析。我们使用这些方法对 122 个人类和小鼠的转录起始位点 cap 分析基因表达 (CAGE) 样本进行分析,构建了包含三个层次的转录起始位点、转录起始簇和转录起始区域的全基因组“启动子组”。