Geller Scott F, Guerin Karen I, Visel Meike, Pham Aaron, Lee Edwin S, Dror Amiel A, Avraham Karen B, Hayashi Toshinori, Ray Catherine A, Reh Thomas A, Bermingham-McDonogh Olivia, Triffo William J, Bao Shaowen, Isosomppi Juha, Västinsalo Hanna, Sankila Eeva-Marja, Flannery John G
Helen Wills Neuroscience Institute, University of California, Berkeley, CA, USA.
PLoS Genet. 2009 Aug;5(8):e1000607. doi: 10.1371/journal.pgen.1000607. Epub 2009 Aug 14.
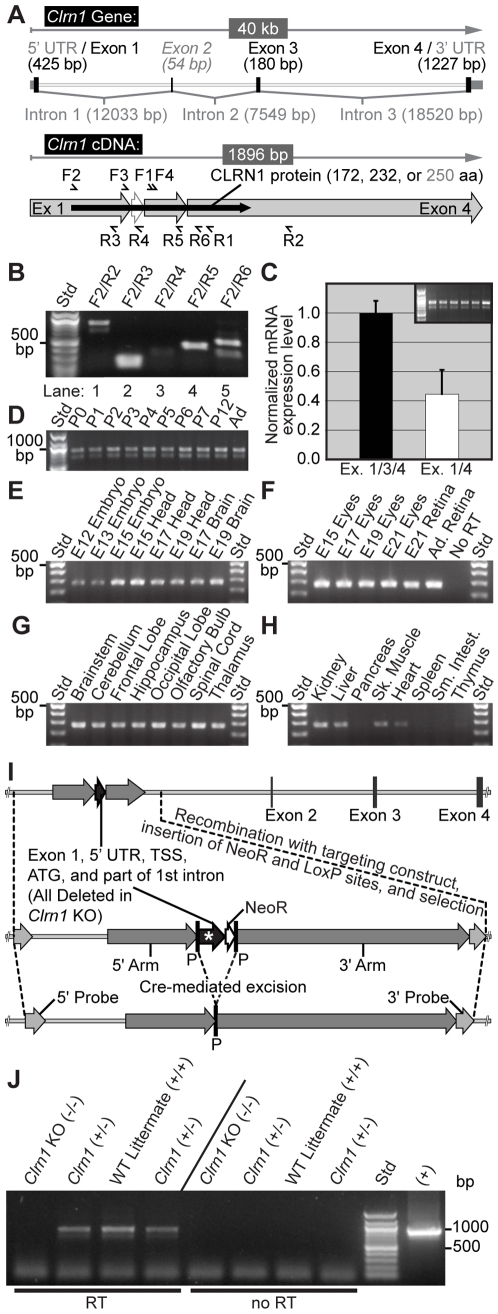
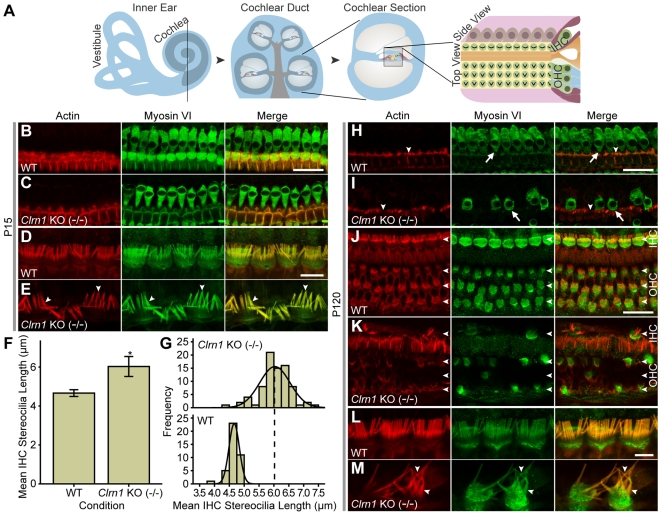
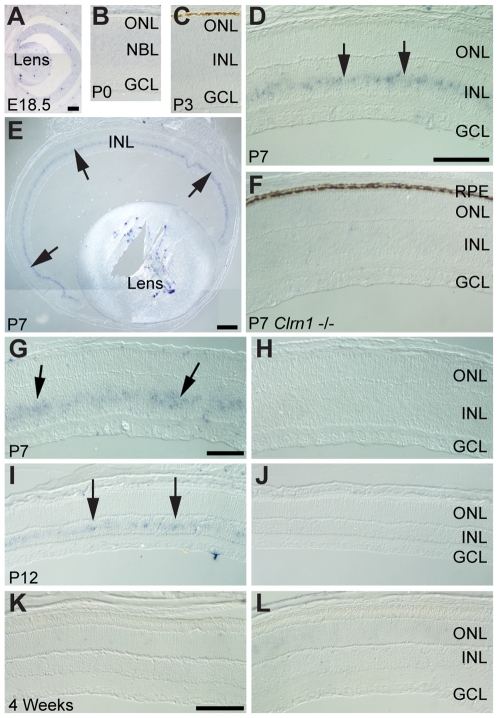
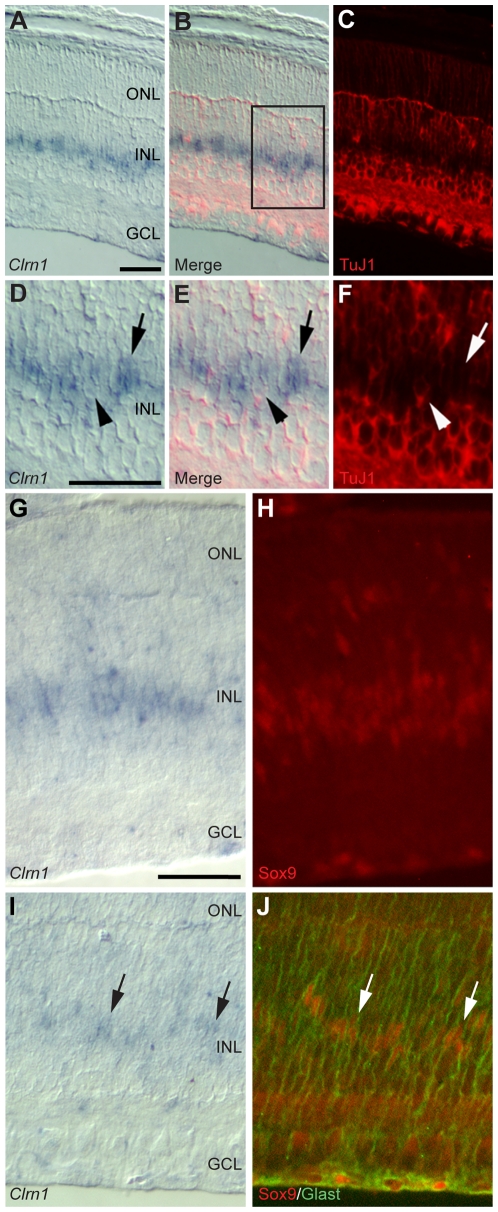
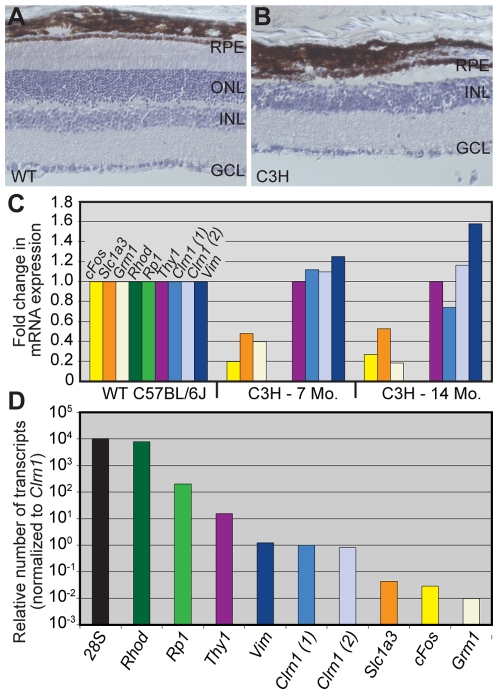
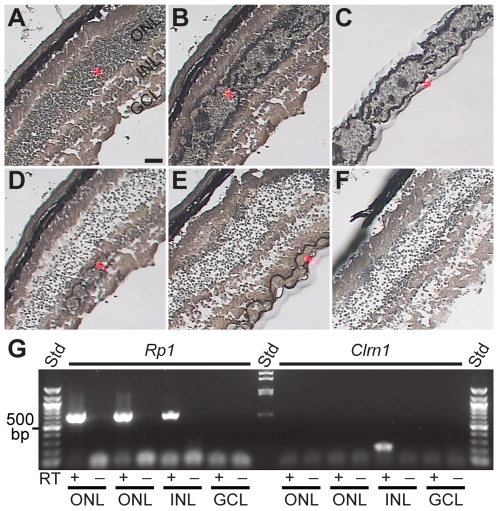
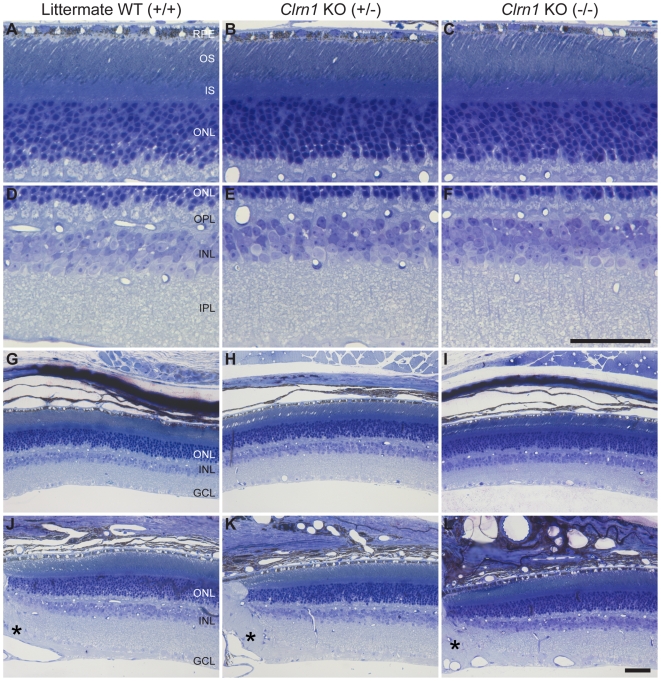
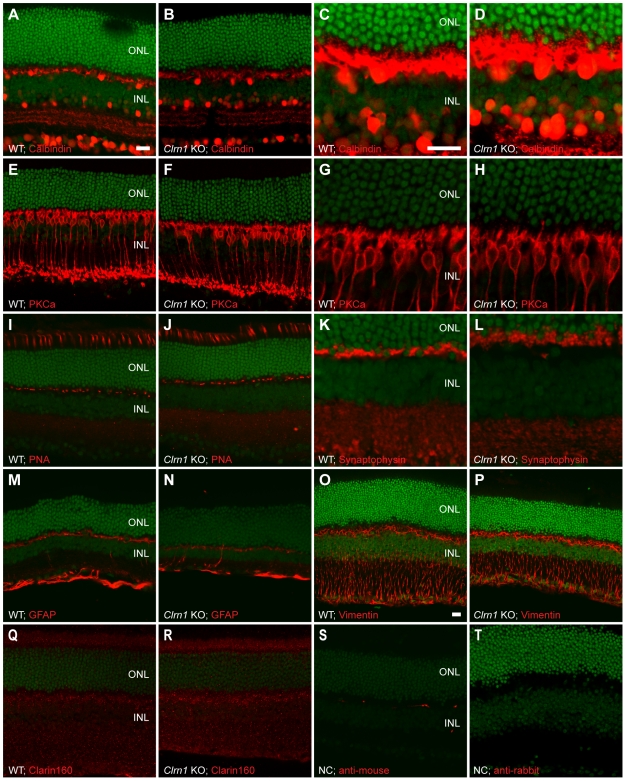
Mutations in the CLRN1 gene cause Usher syndrome type 3 (USH3), a human disease characterized by progressive blindness and deafness. Clarin 1, the protein product of CLRN1, is a four-transmembrane protein predicted to be associated with ribbon synapses of photoreceptors and cochlear hair cells, and recently demonstrated to be associated with the cytoskeleton. To study Clrn1, we created a Clrn1 knockout (KO) mouse and characterized the histological and functional consequences of Clrn1 deletion in the retina and cochlea. Clrn1 KO mice do not develop a retinal degeneration phenotype, but exhibit progressive loss of sensory hair cells in the cochlea and deterioration of the organ of Corti by 4 months. Hair cell stereocilia in KO animals were longer and disorganized by 4 months, and some Clrn1 KO mice exhibited circling behavior by 5-6 months of age. Clrn1 mRNA expression was localized in the retina using in situ hybridization (ISH), laser capture microdissection (LCM), and RT-PCR. Retinal Clrn1 transcripts were found throughout development and adulthood by RT-PCR, although expression peaked at P7 and declined to undetectable levels in adult retina by ISH. LCM localized Clrn1 transcripts to the retinas inner nuclear layer, and WT levels of retinal Clrn1 expression were observed in photoreceptor-less retinas. Examination of Clrn1 KO mice suggests that CLRN1 is unnecessary in the murine retina but essential for normal cochlear development and function. This may reflect a redundancy in the mouse retina not present in human retina. In contrast to mouse KO models of USH1 and USH2, our data indicate that Clrn1 expression in the retina is restricted to the Müller glia. This is a novel finding, as most retinal degeneration associated proteins are expressed in photoreceptors, not in glia. If CLRN1 expression in humans is comparable to the expression pattern observed in mice, this is the first report of an inner retinal protein that, when mutated, causes retinal degeneration.
CLRN1基因突变会导致3型Usher综合征(USH3),这是一种以进行性失明和失聪为特征的人类疾病。Clarin 1是CLRN1的蛋白质产物,是一种四跨膜蛋白,预计与光感受器和耳蜗毛细胞的带状突触相关,最近被证明与细胞骨架有关。为了研究Clrn1,我们创建了一个Clrn1基因敲除(KO)小鼠,并对视网膜和耳蜗中Clrn1缺失的组织学和功能后果进行了表征。Clrn1基因敲除小鼠没有出现视网膜变性表型,但在4个月时耳蜗中的感觉毛细胞逐渐丧失,柯蒂氏器退化。到4个月时,基因敲除动物的毛细胞静纤毛更长且排列紊乱,一些Clrn1基因敲除小鼠在5至6个月大时出现转圈行为。使用原位杂交(ISH)、激光捕获显微切割(LCM)和逆转录聚合酶链反应(RT-PCR)将Clrn1 mRNA表达定位在视网膜中。通过RT-PCR在整个发育和成年期都发现了视网膜Clrn1转录本,尽管其表达在出生后第7天达到峰值,并通过ISH在成年视网膜中降至无法检测的水平。LCM将Clrn1转录本定位到视网膜内核层,并且在无感光细胞的视网膜中观察到视网膜Clrn1表达的野生型水平。对Clrn1基因敲除小鼠的检查表明,CLRN1在小鼠视网膜中不是必需的,但对正常的耳蜗发育和功能至关重要。这可能反映了小鼠视网膜中存在人类视网膜中不存在的冗余现象。与USH1和USH2的小鼠基因敲除模型相比,我们的数据表明Clrn1在视网膜中的表达仅限于米勒胶质细胞。这是一个新发现,因为大多数与视网膜变性相关的蛋白质都在光感受器中表达,而不是在胶质细胞中表达。如果人类中CLRN1的表达与在小鼠中观察到的表达模式相似,这是首次报道一种视网膜内层蛋白质,当其发生突变时会导致视网膜变性。