Ledley F D, Jansen R, Nham S U, Fenton W A, Rosenberg L E
Howard Hughes Medical Institute, Baylor College of Medicine, Houston, TX 77030.
Proc Natl Acad Sci U S A. 1990 Apr;87(8):3147-50. doi: 10.1073/pnas.87.8.3147.
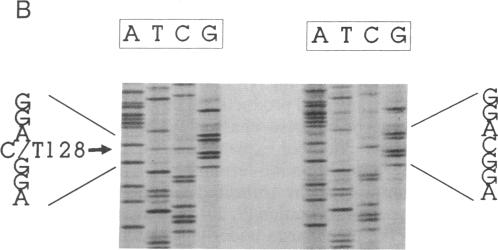
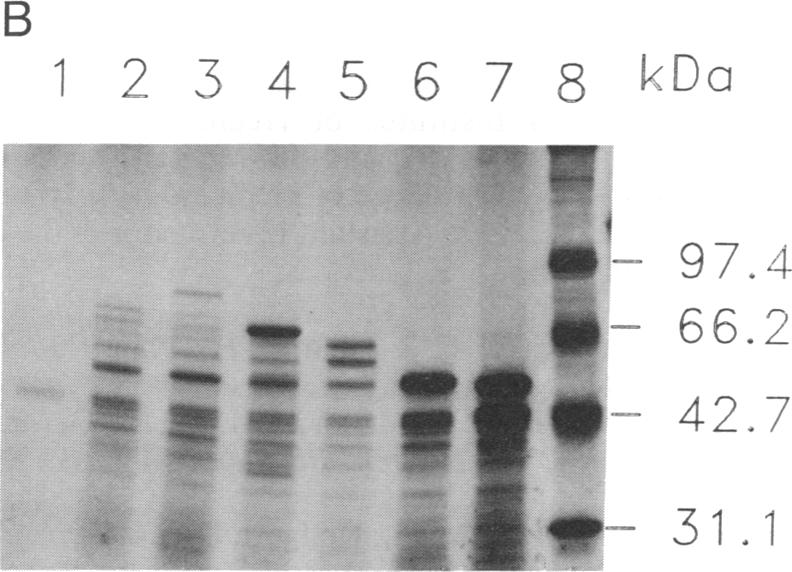
Methylmalonyl-CoA mutase (EC 5.4.99.2) is a mitochondrial matrix enzyme whose activity is deficient in the inherited disorder methylmalonic acidemia. Previous studies on primary fibroblast cell lines from patients with methylmalonic acidemia have delineated a variety of biochemical phenotypes underlying this disorder. One cell line with primary mutase apoenzyme deficiency exhibited a particularly unusual phenotype; it expressed an abnormally small and unstable immunoreactive protein, which was not imported by mitochondria. We now report cloning and sequencing of the cDNA encoding this mutant protein. The mutation is a single base change, a cytosine----thymine transition, which introduces an amber termination codon at position 17 within the mitochondrial leader sequence. The immunoreactive protein produced by these cells reflects translation from AUG codons downstream from this termination codon and, hence, lacks a mitochondrial leader peptide. This mutation represents a complex prototype for a class of mutations in which absence of the mitochondrial targeting sequence leads to absence of a functioning gene product.
甲基丙二酸单酰辅酶A变位酶(EC 5.4.99.2)是一种线粒体基质酶,其活性在遗传性疾病甲基丙二酸血症中缺乏。先前对甲基丙二酸血症患者原代成纤维细胞系的研究已经描绘了这种疾病背后的多种生化表型。一种原发性变位酶脱辅基酶缺乏的细胞系表现出一种特别不寻常的表型;它表达了一种异常小且不稳定的免疫反应性蛋白,该蛋白未被线粒体导入。我们现在报告编码这种突变蛋白的cDNA的克隆和测序。该突变是单个碱基变化,胞嘧啶到胸腺嘧啶的转换,它在线粒体前导序列内的第17位引入了一个琥珀色终止密码子。这些细胞产生的免疫反应性蛋白反映了从该终止密码子下游的AUG密码子进行的翻译,因此缺乏线粒体前导肽。这种突变代表了一类突变的复杂原型,其中线粒体靶向序列的缺失导致功能性基因产物的缺失。