Sabourdy Frédérique, Labauge Pierre, Stensland Hilde Monica Frostad Riise, Nieto Michèle, Garcés Violeta Latorre, Renard Dimitri, Castelnovo Giovanni, de Champfleur Nicolas, Levade Thierry
Laboratoire de Biochimie 'Maladies Métaboliques', Institut Fédératif de Biologie, CHU Purpan, Toulouse, France.
BMC Med Genet. 2009 Sep 3;10:84. doi: 10.1186/1471-2350-10-84.
beta-Mannosidosis (OMIM 248510) is a rare inborn lysosomal storage disorder caused by the deficient activity of beta-mannosidase, an enzyme encoded by a single gene (MANBA) located on chromosome 4q22-25. To date, only 20 cases of this autosomal recessive disorder have been described and 14 different MANBA mutations were incriminated in the disease. These are all null mutations or missense mutations that abolish beta-mannosidase activity. In this study, we characterized the molecular defect of a new case of beta-mannosidosis, presenting with a severe neurological disorder.
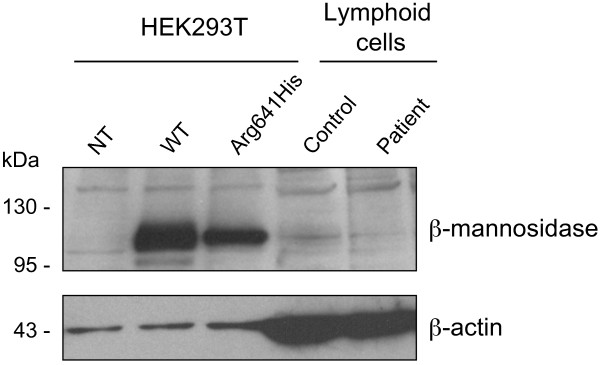
Genomic DNA was isolated from peripheral blood leukocytes of the patient to allow MANBA sequencing. The identified mutation was engineered by site-directed mutagenesis and the mutant protein was expressed through transient transfection in HEK293T cells. The beta-mannosidase expression and activity were respectively assessed by Western blot and fluorometric assay in both leukocytes and HEK293T cells.
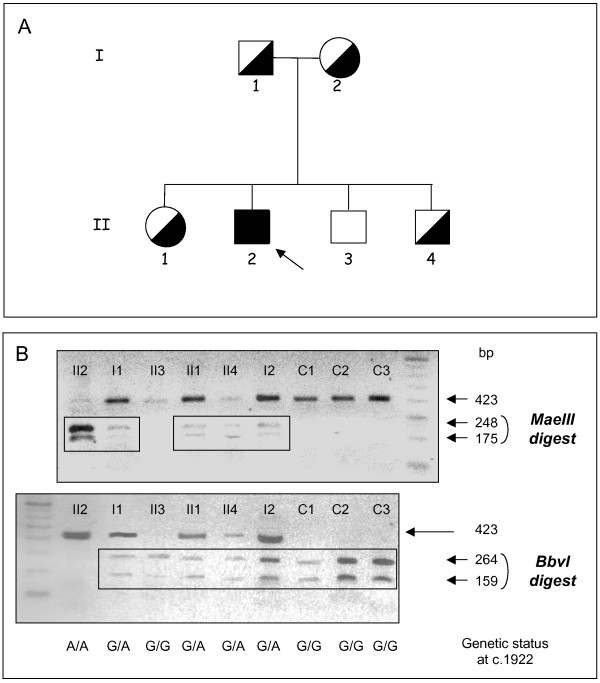
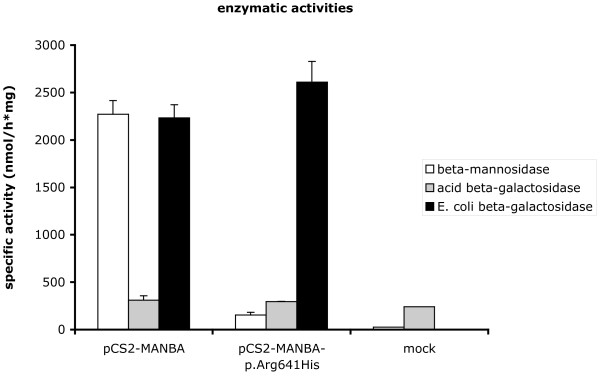
A missense disease-associated mutation, c.1922G>A (p.Arg641His), was identified for which the patient was homozygous. In contrast to previously described missense mutations, this substitution does not totally abrogate the enzyme activity but led to a residual activity of about 7% in the patient's leukocytes, 11% in lymphoblasts and 14% in plasma. Expression studies in transfected cells also resulted in 7% residual activity.
Correlations between MANBA mutations, residual activity of beta-mannosidase and the severity of the ensuing neurological disorder are discussed. Whether the c.1922G>A mutation is responsible for a yet undescribed pseudodeficiency of beta-mannosidase is also discussed.
β-甘露糖苷贮积症(OMIM 248510)是一种罕见的先天性溶酶体贮积病,由β-甘露糖苷酶活性缺乏引起,该酶由位于4q22 - 25染色体上的单基因(MANBA)编码。迄今为止,仅报道了20例这种常染色体隐性疾病病例,并且已确定14种不同的MANBA突变与该疾病相关。这些均为无义突变或错义突变,可消除β-甘露糖苷酶活性。在本研究中,我们对1例表现为严重神经疾病的β-甘露糖苷贮积症新病例的分子缺陷进行了特征分析。
从患者外周血白细胞中分离基因组DNA以进行MANBA测序。通过定点诱变构建鉴定出的突变,并通过在HEK293T细胞中瞬时转染表达突变蛋白。分别通过蛋白质免疫印迹法和荧光测定法评估白细胞和HEK293T细胞中β-甘露糖苷酶的表达和活性。
鉴定出一种错义疾病相关突变c.1922G>A(p.Arg641His),患者为该突变的纯合子。与先前描述的错义突变不同,这种替代并未完全消除酶活性,但导致患者白细胞中约7%的残余活性,淋巴细胞中为11%,血浆中为14%。转染细胞中的表达研究也产生了7%的残余活性。
讨论了MANBA突变、β-甘露糖苷酶残余活性与随后神经疾病严重程度之间的相关性。还讨论了c.1922G>A突变是否导致尚未描述的β-甘露糖苷酶假缺陷。