Lahti Jennifer L, Silverman Adam P, Cochran Jennifer R
Department of Bioengineering, Cancer Center, Bio-X Program, Stanford University, Stanford, California, United States of America.
PLoS Comput Biol. 2009 Sep;5(9):e1000499. doi: 10.1371/journal.pcbi.1000499. Epub 2009 Sep 4.
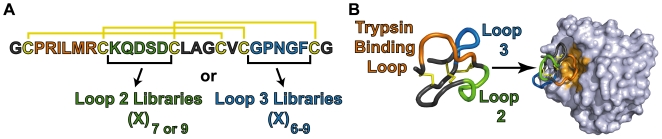
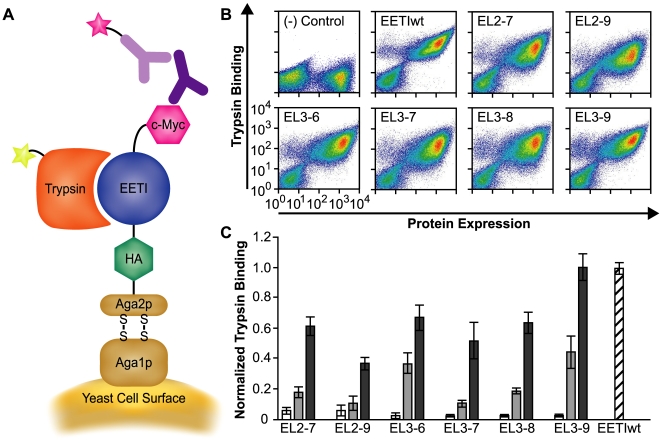
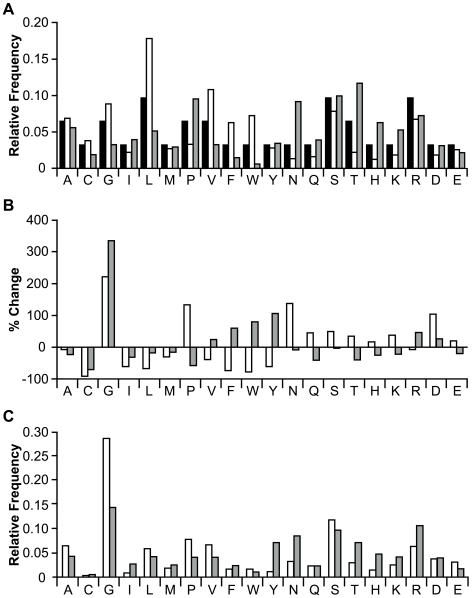
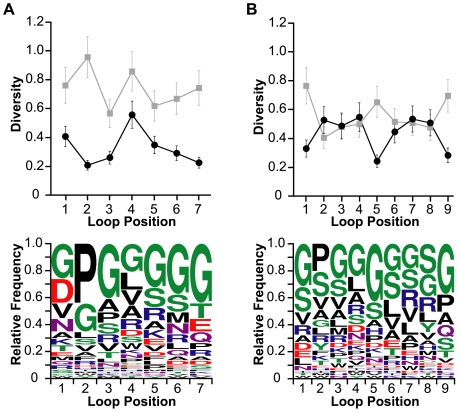
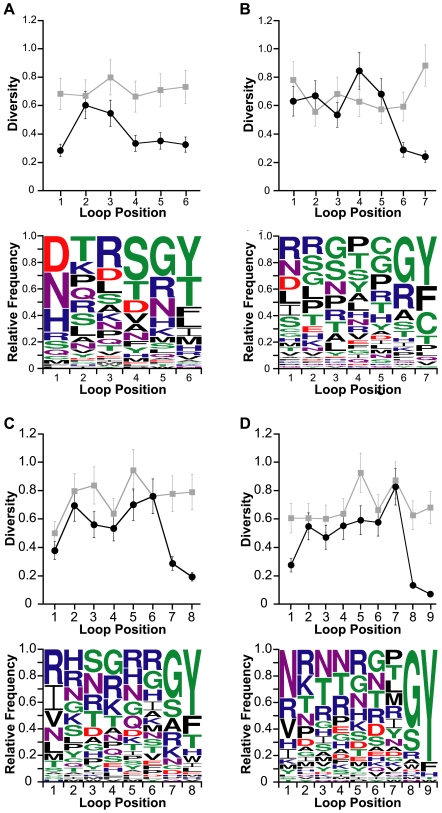
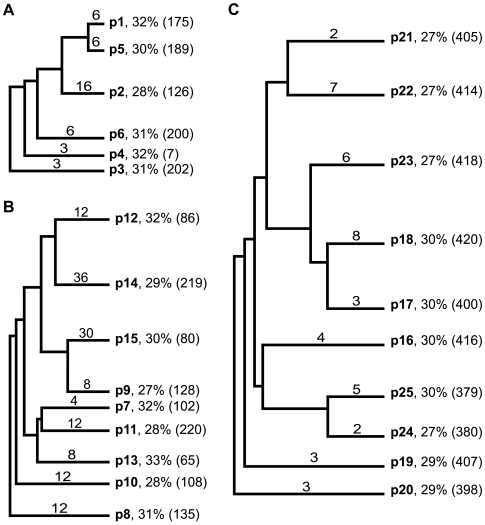
Cystine-knot miniproteins (knottins) are promising molecular scaffolds for protein engineering applications. Members of the knottin family have multiple loops capable of displaying conformationally constrained polypeptides for molecular recognition. While previous studies have illustrated the potential of engineering knottins with modified loop sequences, a thorough exploration into the tolerated loop lengths and sequence space of a knottin scaffold has not been performed. In this work, we used the Ecballium elaterium trypsin inhibitor II (EETI) as a model member of the knottin family and constructed libraries of EETI loop-substituted variants with diversity in both amino acid sequence and loop length. Using yeast surface display, we isolated properly folded EETI loop-substituted clones and applied sequence analysis tools to assess the tolerated diversity of both amino acid sequence and loop length. In addition, we used covariance analysis to study the relationships between individual positions in the substituted loops, based on the expectation that correlated amino acid substitutions will occur between interacting residue pairs. We then used the results of our sequence and covariance analyses to successfully predict loop sequences that facilitated proper folding of the knottin when substituted into EETI loop 3. The sequence trends we observed in properly folded EETI loop-substituted clones will be useful for guiding future protein engineering efforts with this knottin scaffold. Furthermore, our findings demonstrate that the combination of directed evolution with sequence and covariance analyses can be a powerful tool for rational protein engineering.
胱氨酸结微型蛋白(结蛋白)是蛋白质工程应用中很有前景的分子支架。结蛋白家族成员有多个环,能够展示构象受限的多肽用于分子识别。虽然先前的研究已经阐明了通过修饰环序列来改造结蛋白的潜力,但尚未对结蛋白支架可耐受的环长度和序列空间进行全面探索。在这项工作中,我们使用栝楼胰蛋白酶抑制剂II(EETI)作为结蛋白家族的一个模型成员,并构建了EETI环取代变体文库,该文库在氨基酸序列和环长度上都具有多样性。利用酵母表面展示技术,我们分离出了正确折叠的EETI环取代克隆,并应用序列分析工具来评估氨基酸序列和环长度的可耐受多样性。此外,基于相互作用残基对之间会发生相关氨基酸取代的预期,我们使用协方差分析来研究取代环中各个位置之间的关系。然后,我们利用序列和协方差分析的结果成功预测了在取代EETI环3时有助于结蛋白正确折叠的环序列。我们在正确折叠的EETI环取代克隆中观察到的序列趋势将有助于指导未来使用这种结蛋白支架进行的蛋白质工程工作。此外,我们的研究结果表明,定向进化与序列和协方差分析相结合可以成为合理进行蛋白质工程的有力工具。