Department of Physiology and Pharmacology, University of Strathclyde, Strathclyde Institute for Pharmacy and Biomedical Sciences, Glasgow, Scotland, UK.
Cell Signal. 2010 Feb;22(2):265-73. doi: 10.1016/j.cellsig.2009.09.028. Epub 2009 Sep 23.
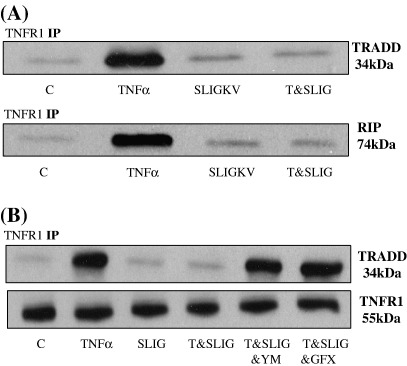
In this study we examined the potential for PAR(2) and TNFalpha to synergise at the level of MAP kinase signalling in PAR(2) expressing NCTC2544 cells. However, to our surprise we found that activation of PAR(2) by trypsin or the specific activating peptide SLIGKV-OH strongly inhibited both the phosphorylation and activity of JNK. In contrast neither p38 MAP kinase nor ERK activation was affected although TNFalpha stimulated IkappaBalpha loss was partially reversed. The inhibitory effect was not observed in parental cells nor in cells expressing PAR(4), however inhibition was reversed by pre-incubation with the novel PAR(2) antagonist K14585, suggesting that the effect is specific for PAR(2) activation. SLIGKV-OH was found to be more potent in inhibiting TNFalpha-induced JNK activation than in stimulating JNK alone, suggesting agonist-directed signalling. The PKC activator PMA, also mimicked the inhibitory effect of SLIGKV-OH, and the effects of both agents were reversed by pre-treatment with the PKC inhibitor, GF109203X. Furthermore, incubation with the novel G(q/11) inhibitor YM25480 also reversed PAR(2) mediated inhibition. Activation of PAR(2) was found to disrupt TNFR1 binding to RIP and TRADD and this was reversed by both GF109203X and YM25480. A similar mode of inhibition observed in HUVECs through PAR(2) or P2Y2 receptors demonstrates the potential of a novel paradigm for GPCRs linked to G(q/11), in mediating inhibition of TNFalpha-stimulated JNK activation. This has important implications in assessing the role of GPCRs in inflammation and other conditions.
在这项研究中,我们研究了 PAR(2)和 TNFalpha 在 PAR(2)表达的 NCTC2544 细胞中 MAP 激酶信号传导水平上协同作用的潜力。然而,令我们惊讶的是,我们发现胰蛋白酶或特异性激活肽 SLIGKV-OH 激活 PAR(2)强烈抑制 JNK 的磷酸化和活性。相比之下,p38 MAP 激酶或 ERK 的激活不受影响,尽管 TNFalpha 刺激 IkappaBalpha 的丢失部分得到逆转。这种抑制作用在亲本细胞或表达 PAR(4)的细胞中均未观察到,但是在用新型 PAR(2)拮抗剂 K14585 预处理后,抑制作用被逆转,这表明该作用是 PAR(2)激活的特异性作用。发现 SLIGKV-OH 抑制 TNFalpha 诱导的 JNK 激活比单独刺激 JNK 更有效,提示激动剂导向的信号传导。PKC 激活剂 PMA 也模拟了 SLIGKV-OH 的抑制作用,并且两种试剂的作用都可以通过用 PKC 抑制剂 GF109203X 预处理来逆转。此外,用新型 G(q/11)抑制剂 YM25480 孵育也逆转了 PAR(2)介导的抑制。发现 PAR(2)的激活破坏了 TNFR1 与 RIP 和 TRADD 的结合,这两种结合均被 GF109203X 和 YM25480 逆转。通过 PAR(2)或 P2Y2 受体在 HUVECs 中观察到的类似抑制模式表明,一种新的 GPCR 与 G(q/11)相关联的范式,在介导 TNFalpha 刺激的 JNK 激活的抑制中具有潜力。这对于评估 GPCR 在炎症和其他情况下的作用具有重要意义。