Austin Eric D, Phillips John A, Cogan Joy D, Hamid Rizwan, Yu Chang, Stanton Krista C, Phillips Charles A, Wheeler Lisa A, Robbins Ivan M, Newman John H, Loyd James E
Department of Pediatrics, Vanderbilt University, Medical Center, Nashville, TN, USA.
Respir Res. 2009 Sep 28;10(1):87. doi: 10.1186/1465-9921-10-87.
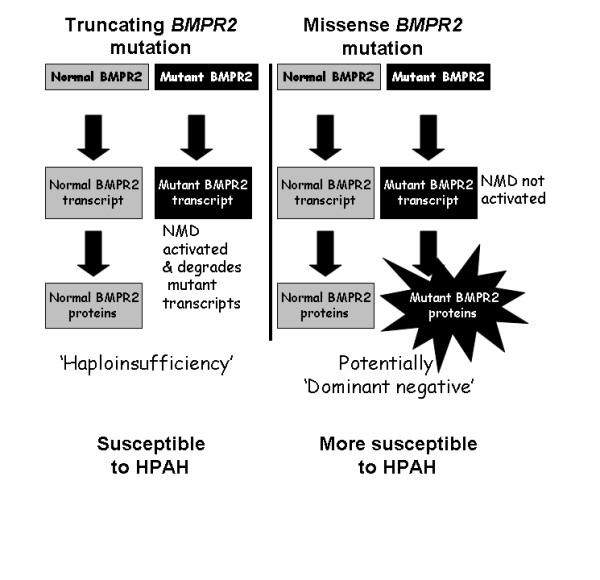
Autosomal dominant inheritance of germline mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene are a major risk factor for pulmonary arterial hypertension (PAH). While previous studies demonstrated a difference in severity between BMPR2 mutation carriers and noncarriers, it is likely disease severity is not equal among BMPR2 mutations. We hypothesized that patients with missense BMPR2 mutations have more severe disease than those with truncating mutations.
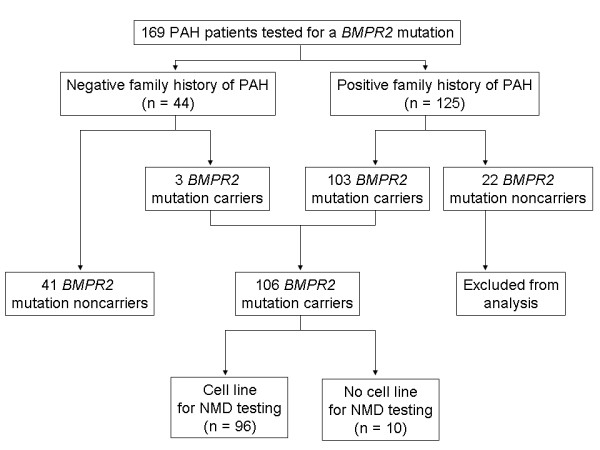
Testing for BMPR2 mutations was performed in 169 patients with PAH (125 with a family history of PAH and 44 with sporadic disease). Of the 106 patients with a detectable BMPR2 mutation, lymphocytes were available in 96 to functionally assess the nonsense-mediated decay pathway of RNA surveillance. Phenotypic characteristics were compared between BMPR2 mutation carriers and noncarriers, as well as between those carriers with a missense versus truncating mutation.
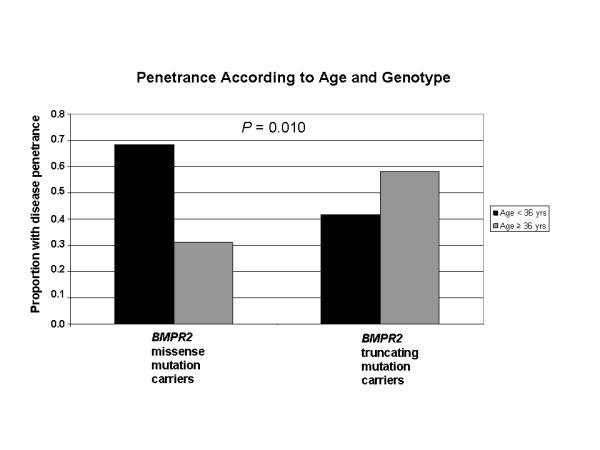
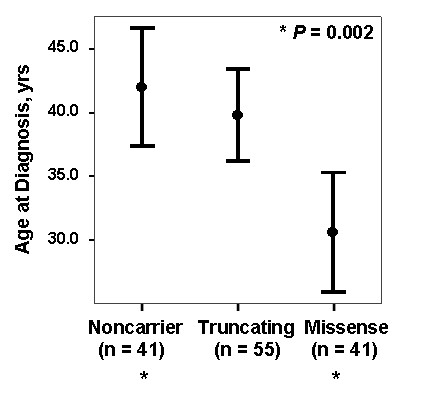
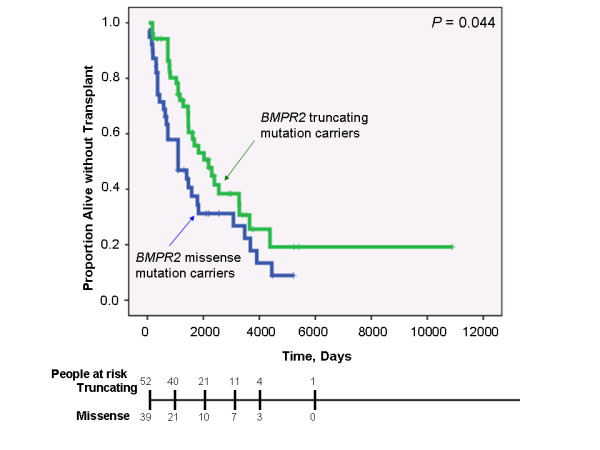
While there was a statistically significant difference in age at diagnosis between carriers and noncarriers, subgroup analysis revealed this to be the case only for females. Among carriers, there was no difference in age at diagnosis, death, or survival according to exonic location of the BMPR2 mutation. However, patients with missense mutations had statistically significant younger ages at diagnosis and death, as well as shorter survival from diagnosis to death or lung transplantation than those with truncating mutations. Consistent with this data, the majority of missense mutations were penetrant prior to age 36 years, while the majority of truncating mutations were penetrant after age 36 years.
In this cohort, BMPR2 mutation carriers have more severe PAH disease than noncarriers, but this is only the case for females. Among carriers, patients with missense mutations that escape nonsense-mediated decay have more severe disease than those with truncating mutations. These findings suggest that treatment and prevention strategies directed specifically at BMPR2 pathway defects may need to vary according to the type of mutation.
骨形态发生蛋白受体2(BMPR2)基因种系突变的常染色体显性遗传是肺动脉高压(PAH)的主要危险因素。虽然先前的研究表明BMPR2突变携带者与非携带者在疾病严重程度上存在差异,但BMPR2突变之间的疾病严重程度可能并不相同。我们推测,与截短突变患者相比,错义BMPR2突变患者的疾病更为严重。
对169例PAH患者(125例有PAH家族史,44例为散发性疾病)进行BMPR2突变检测。在106例可检测到BMPR2突变的患者中,96例患者有淋巴细胞可用于功能评估RNA监测的无义介导衰变途径。比较了BMPR2突变携带者与非携带者之间以及错义突变与截短突变携带者之间的表型特征。
虽然携带者与非携带者在诊断年龄上存在统计学显著差异,但亚组分析显示仅女性存在这种情况。在携带者中,根据BMPR2突变的外显子位置,诊断年龄、死亡或生存率没有差异。然而,与截短突变患者相比,错义突变患者在诊断和死亡时的年龄在统计学上显著更小,从诊断到死亡或肺移植的生存期也更短。与该数据一致的是,大多数错义突变在36岁之前具有外显率,而大多数截短突变在36岁之后具有外显率。
在该队列中,BMPR2突变携带者的PAH疾病比非携带者更严重,但仅女性如此。在携带者中,逃避无义介导衰变的错义突变患者比截短突变患者的疾病更严重。这些发现表明,针对BMPR2途径缺陷的治疗和预防策略可能需要根据突变类型而有所不同。