Hikita Hayato, Takehara Tetsuo, Kodama Takahiro, Shimizu Satoshi, Hosui Atsushi, Miyagi Takuya, Tatsumi Tomohide, Ishida Hisashi, Ohkawa Kazuyoshi, Li Wei, Kanto Tatsuya, Hiramatsu Naoki, Hennighausen Lothar, Yin Xiao-Ming, Hayashi Norio
Department of Gastroenterology and Hepatology, Osaka University Graduate School of Medicine, Osaka, Japan.
Hepatology. 2009 Dec;50(6):1972-80. doi: 10.1002/hep.23207.
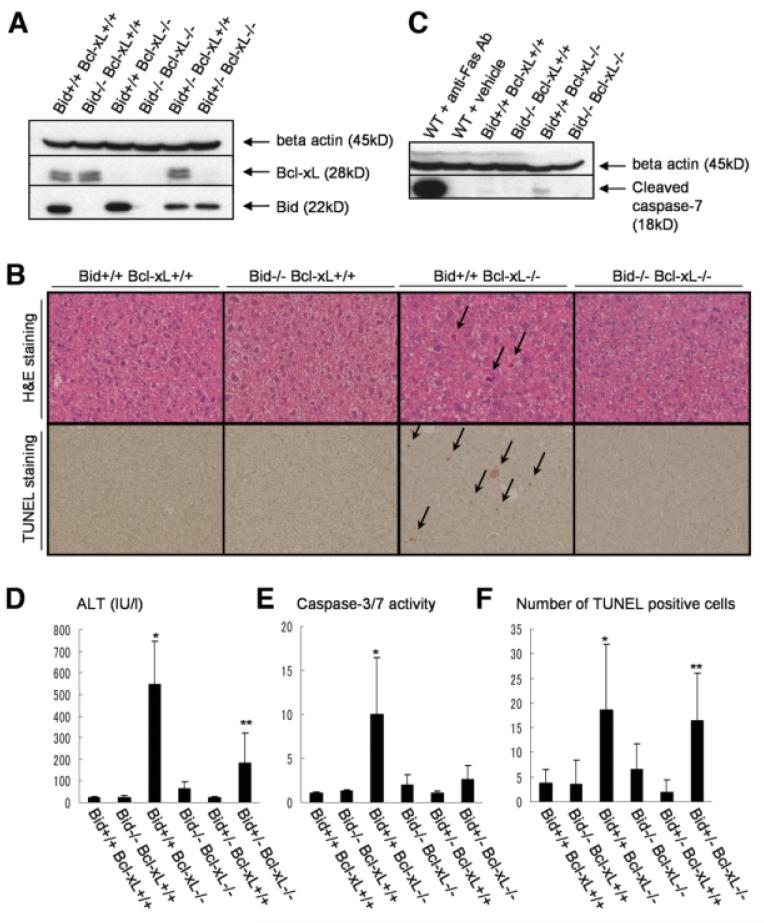
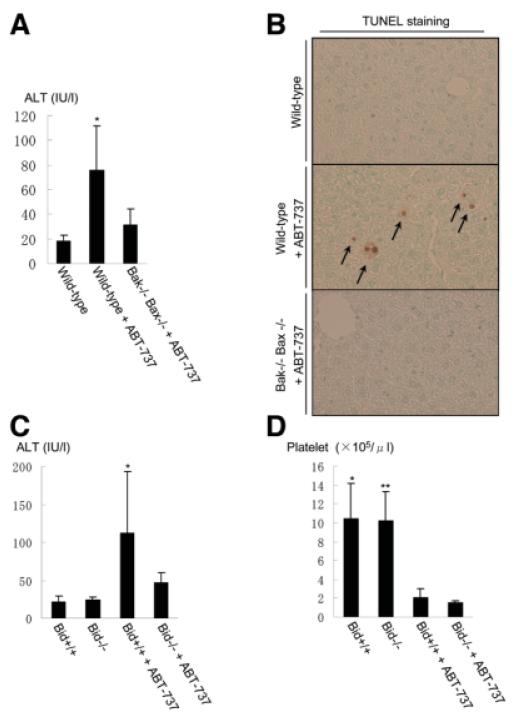
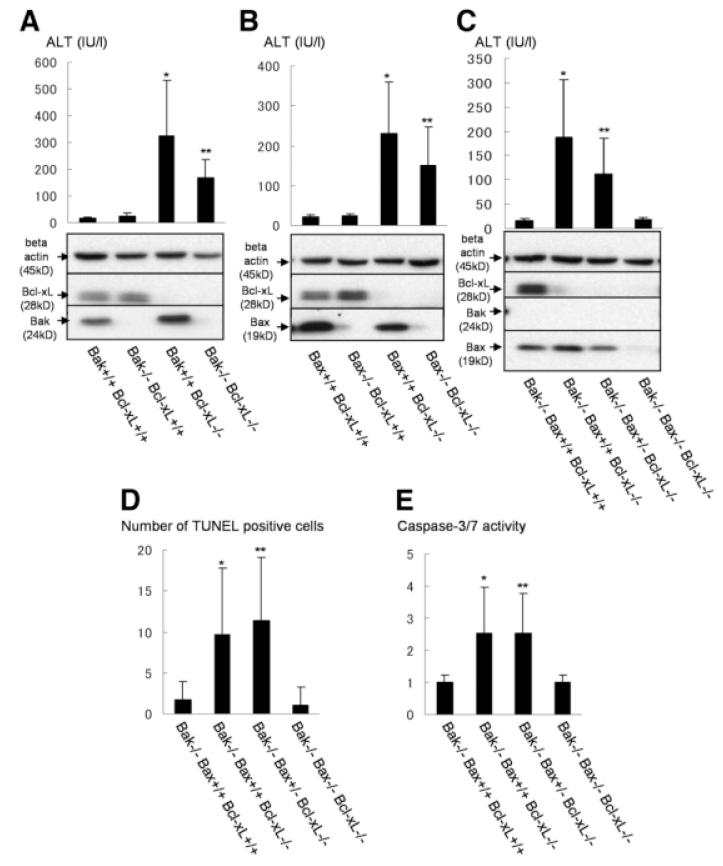
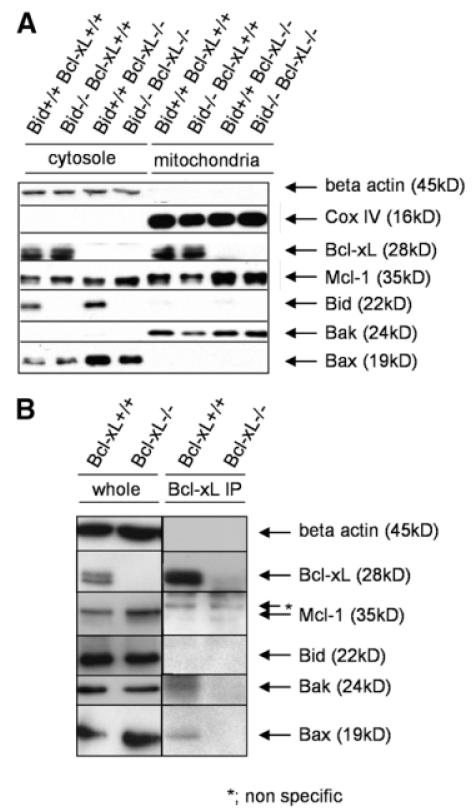
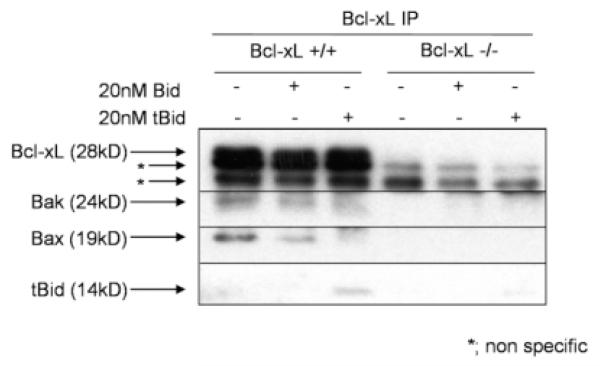
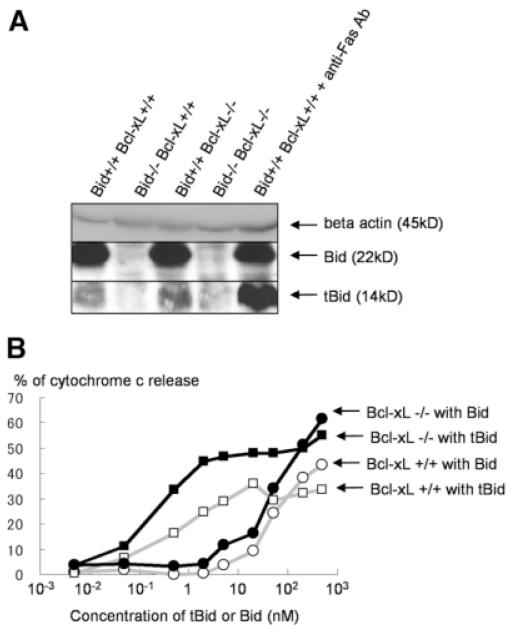
Bcl-2 homology domain 3 (BH3)-only protein Bid is posttranslationally cleaved by caspase-8 into its truncated form (tBid) and couples with stress signals to the mitochondrial cell death pathway. However, the physiological relevance of Bid is not clearly understood. Hepatocyte-specific knockout (KO) of Bcl-xL leads to naturally-occurring apoptosis despite co-expression of Mcl-1, which shares a similar anti-apoptotic function. We generated Bcl-xL KO, Bcl-xL/Bid double KO, Bcl-xL/Bak double KO, Bcl-xL/Bax double KO, and Bcl-xL/Bak/Bax triple KO mice and found that hepatocyte apoptosis caused by Bcl-xL deficiency was completely dependent on Bak and Bax, and surprisingly on Bid. This indicated that, in the absence of Bid, Bcl-xL is not required for the integrity of differentiated hepatocytes, suggesting a complicated interaction between core Bcl-2 family proteins and BH3-only proteins even in a physiological setting. Indeed, a small but significant level of tBid was present in wild-type liver under physiological conditions. tBid was capable of binding to Bcl-xL and displacing Bak and Bax from Bcl-xL, leading to release of cytochrome c from wild-type mitochondria. Bcl-xL-deficient mitochondria were more susceptible to tBid-induced cytochrome c release. Finally, administration of ABT-737, a pharmacological inhibitor of Bcl-2/Bcl-xL, caused Bak/Bax-dependent liver injury, but this was clearly ameliorated with a Bid KO background.
Bid, originally considered to be a sensor for apoptotic stimuli, is constitutively active in healthy liver cells and is involved in the Bak/Bax-dependent mitochondrial cell death pathway. Healthy liver cells are addicted to a single Bcl-2-like molecule because of BH3 stresses, and therefore special caution may be required for the use of the Bcl-2 inhibitor for cancer therapy.
仅含Bcl-2同源结构域3(BH3)的蛋白Bid在翻译后被半胱天冬酶-8切割成截短形式(tBid),并将应激信号与线粒体细胞死亡途径偶联。然而,Bid的生理相关性尚不清楚。尽管共表达具有相似抗凋亡功能的Mcl-1,但Bcl-xL的肝细胞特异性敲除(KO)仍会导致自然发生的细胞凋亡。我们构建了Bcl-xL基因敲除、Bcl-xL/Bid双基因敲除、Bcl-xL/Bak双基因敲除、Bcl-xL/Bax双基因敲除以及Bcl-xL/Bak/Bax三基因敲除小鼠,发现Bcl-xL缺乏引起的肝细胞凋亡完全依赖于Bak和Bax,且令人惊讶地依赖于Bid。这表明,在缺乏Bid的情况下,分化肝细胞的完整性不需要Bcl-xL,这表明即使在生理环境中,核心Bcl-2家族蛋白与仅含BH3的蛋白之间也存在复杂的相互作用。事实上,在生理条件下野生型肝脏中存在少量但显著水平的tBid。tBid能够与Bcl-xL结合,并将Bak和Bax从Bcl-xL上置换下来,导致野生型线粒体释放细胞色素c。缺乏Bcl-xL的线粒体对tBid诱导的细胞色素c释放更敏感。最后,给予Bcl-2/Bcl-xL的药理学抑制剂ABT-737会导致Bak/Bax依赖性肝损伤,但在Bid基因敲除背景下这种损伤明显减轻。
Bid最初被认为是凋亡刺激的传感器,在健康肝细胞中具有组成性活性,并参与Bak/Bax依赖性线粒体细胞死亡途径。由于BH3应激,健康肝细胞依赖于单一的Bcl-2样分子,因此在癌症治疗中使用Bcl-2抑制剂时可能需要特别谨慎。