Department of Computational Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Bioinformatics. 2010 Feb 1;26(3):319-25. doi: 10.1093/bioinformatics/btp664. Epub 2009 Dec 4.
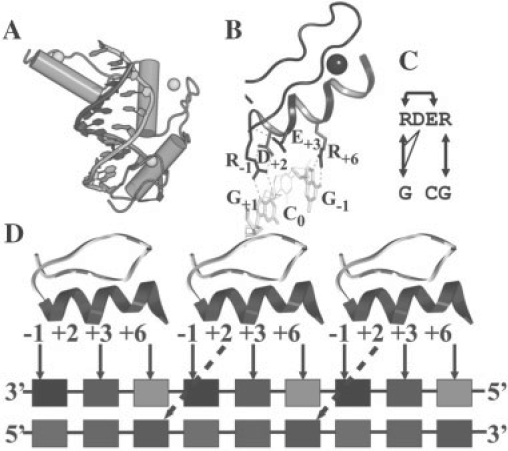
A major limitation in modeling protein interactions is the difficulty of assessing the over-fitting of the training set. Recently, an experimentally based approach that integrates crystallographic information of C2H2 zinc finger-DNA complexes with binding data from 11 mutants, 7 from EGR finger I, was used to define an improved interaction code (no optimization). Here, we present a novel mixed integer programming (MIP)-based method that transforms this type of data into an optimized code, demonstrating both the advantages of the mathematical formulation to minimize over- and under-fitting and the robustness of the underlying physical parameters mapped by the code.
Based on the structural models of feasible interaction networks for 35 mutants of EGR-DNA complexes, the MIP method minimizes the cumulative binding energy over all complexes for a general set of fundamental protein-DNA interactions. To guard against over-fitting, we use the scalability of the method to probe against the elimination of related interactions. From an initial set of 12 parameters (six hydrogen bonds, five desolvation penalties and a water factor), we proceed to eliminate five of them with only a marginal reduction of the correlation coefficient to 0.9983. Further reduction of parameters negatively impacts the performance of the code (under-fitting). Besides accurately predicting the change in binding affinity of validation sets, the code identifies possible context-dependent effects in the definition of the interaction networks. Yet, the approach of constraining predictions to within a pre-selected set of interactions limits the impact of these potential errors to related low-affinity complexes.
Supplementary data are available at Bioinformatics online.
在建模蛋白质相互作用时,一个主要的限制是难以评估训练集的过拟合。最近,一种基于实验的方法,该方法整合了 C2H2 锌指-DNA 复合物的晶体学信息和来自 11 个突变体的结合数据,其中 7 个来自 EGR 手指 I,用于定义改进的相互作用代码(无优化)。在这里,我们提出了一种新的基于混合整数规划(MIP)的方法,该方法将这种类型的数据转化为优化代码,展示了数学公式在最小化过拟合和欠拟合方面的优势,以及代码所映射的基础物理参数的稳健性。
基于 35 个 EGR-DNA 复合物突变体的可行相互作用网络的结构模型,MIP 方法最小化了所有复合物的累积结合能,适用于一组基本的蛋白质-DNA 相互作用。为了防止过拟合,我们使用该方法的可扩展性来探测相关相互作用的消除。从一组初始的 12 个参数(六个氢键、五个去溶剂化罚分和一个水因子)开始,我们继续消除其中的五个,而相关系数仅略有下降,降至 0.9983。进一步减少参数会对代码的性能产生负面影响(欠拟合)。除了准确预测验证集的结合亲和力变化外,该代码还确定了相互作用网络定义中的可能上下文相关效应。然而,将预测约束在预先选择的相互作用集内的方法限制了这些潜在错误对相关低亲和力复合物的影响。
补充数据可在生物信息学在线获得。