The Fish Molecular Genetics and Biotechnology Laboratory, Department of Fisheries and Allied Aquacultures and Program of Cell and Molecular Biosciences, Aquatic Genomics Unit, Auburn University, Auburn, AL 36849, USA.
BMC Genomics. 2009 Dec 10;10:592. doi: 10.1186/1471-2164-10-592.
Comparative mapping is a powerful tool to transfer genomic information from sequenced genomes to closely related species for which whole genome sequence data are not yet available. However, such an approach is still very limited in catfish, the most important aquaculture species in the United States. This project was initiated to generate additional BAC end sequences and demonstrate their applications in comparative mapping in catfish.
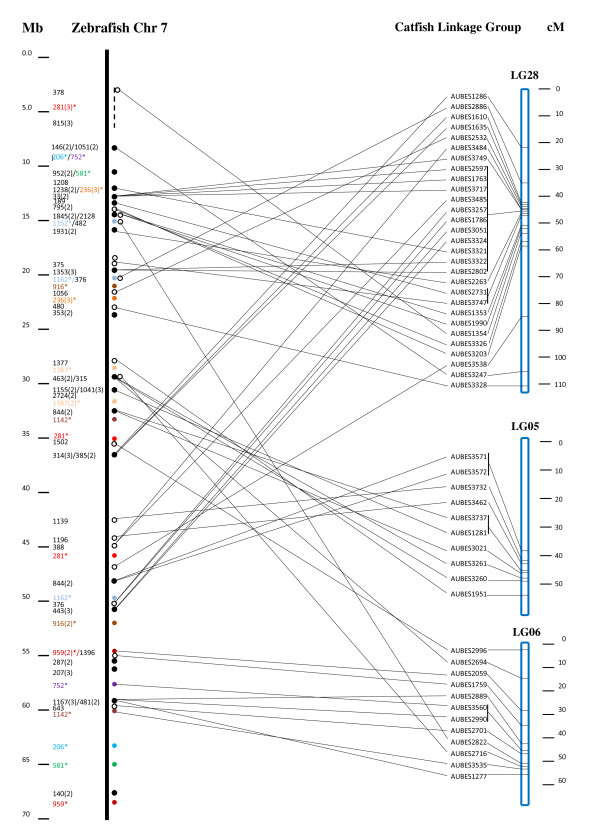
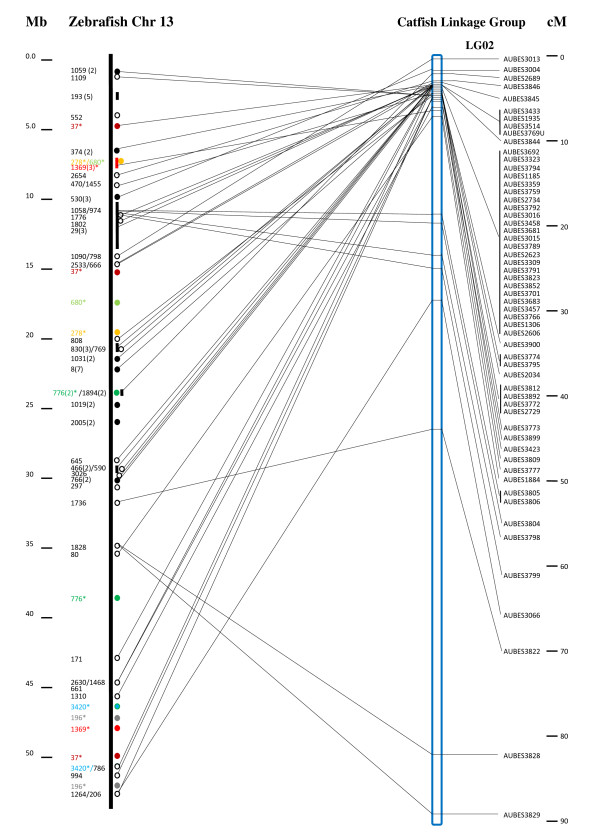
We reported the generation of 43,000 BAC end sequences and their applications for comparative genome analysis in catfish. Using these and the additional 20,000 existing BAC end sequences as a resource along with linkage mapping and existing physical map, conserved syntenic regions were identified between the catfish and zebrafish genomes. A total of 10,943 catfish BAC end sequences (17.3%) had significant BLAST hits to the zebrafish genome (cutoff value <or= e(-5)), of which 3,221 were unique gene hits, providing a platform for comparative mapping based on locations of these genes in catfish and zebrafish. Genetic linkage mapping of microsatellites associated with contigs allowed identification of large conserved genomic segments and construction of super scaffolds.
BAC end sequences and their associated polymorphic markers are great resources for comparative genome analysis in catfish. Highly conserved chromosomal regions were identified to exist between catfish and zebrafish. However, it appears that the level of conservation at local genomic regions are high while a high level of chromosomal shuffling and rearrangements exist between catfish and zebrafish genomes. Orthologous regions established through comparative analysis should facilitate both structural and functional genome analysis in catfish.
比较作图是一种强大的工具,可将基因组信息从已测序的基因组转移到与近缘物种,这些物种尚未获得全基因组序列数据。然而,这种方法在鲶鱼中仍然非常有限,鲶鱼是美国最重要的水产养殖物种。本项目旨在生成更多的 BAC 末端序列,并展示其在鲶鱼比较作图中的应用。
我们报告了生成 43000 个 BAC 末端序列及其在鲶鱼比较基因组分析中的应用。利用这些以及另外 20000 个现有的 BAC 末端序列作为资源,结合连锁作图和现有的物理图谱,在鲶鱼和斑马鱼基因组之间鉴定出保守的同线性区域。共有 10943 个鲶鱼 BAC 末端序列(17.3%)与斑马鱼基因组有显著的 BLAST 命中(截断值 <= e(-5)),其中 3221 个是独特的基因命中,为基于这些基因在鲶鱼和斑马鱼中的位置进行比较作图提供了一个平台。与 contigs 相关的微卫星的遗传连锁作图允许鉴定出大的保守基因组片段,并构建超级支架。
BAC 末端序列及其相关的多态性标记是鲶鱼比较基因组分析的重要资源。在鲶鱼和斑马鱼之间鉴定出高度保守的染色体区域。然而,似乎在局部基因组区域的保守水平很高,而在鲶鱼和斑马鱼基因组之间存在高水平的染色体重排和重组。通过比较分析建立的同源区域应该有助于鲶鱼的结构和功能基因组分析。