Phenomics and Bioinformatics Research Centre, School of Mathematics and Statistics, and Australian Centre for Plant Functional Genomics, University of South Australia, Mawson Lakes Boulevard, Mawson Lakes, SA 5095, Australia.
Genome Biol. 2009;10(12):R142. doi: 10.1186/gb-2009-10-12-r142. Epub 2009 Dec 22.
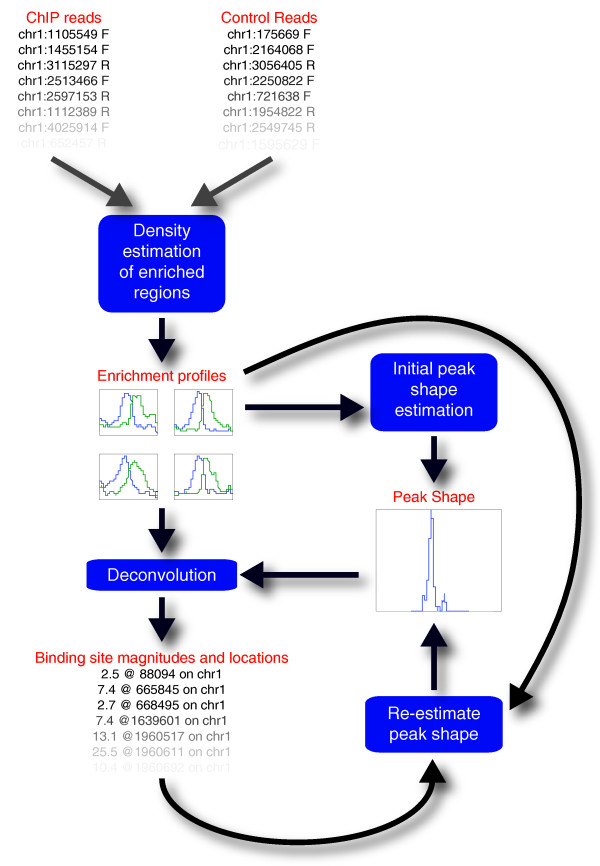
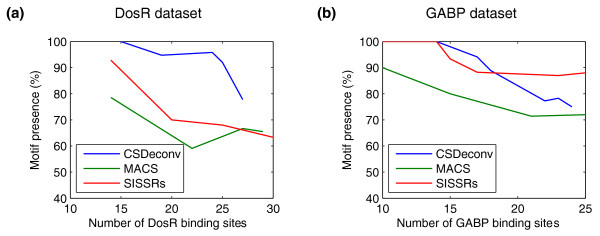
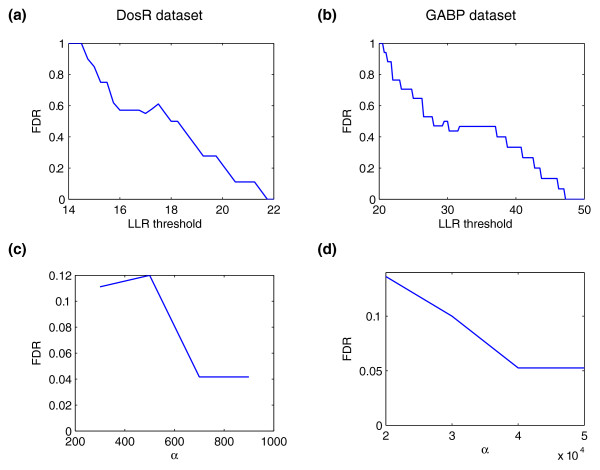
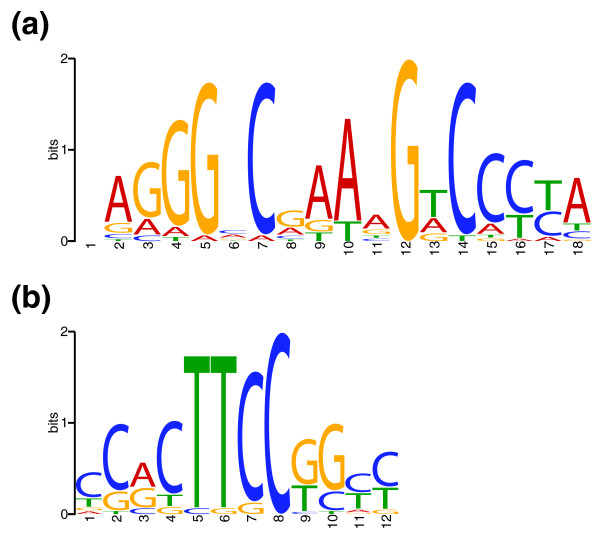
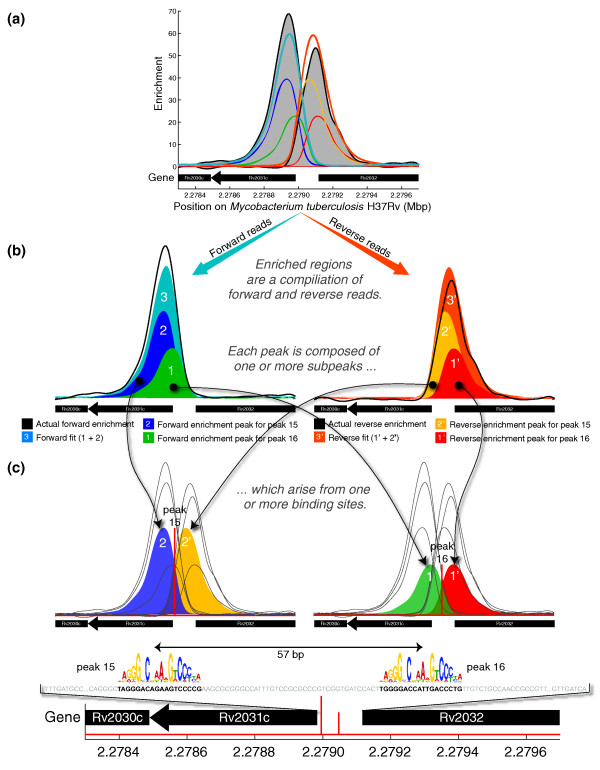
We present CSDeconv, a computational method that determines locations of transcription factor binding from ChIP-seq data. CSDeconv differs from prior methods in that it uses a blind deconvolution approach that allows closely-spaced binding sites to be called accurately. We apply CSDeconv to novel ChIP-seq data for DosR binding in Mycobacterium tuberculosis and to existing data for GABP in humans and show that it can discriminate binding sites separated by as few as 40 bp.
我们提出了 CSDeconv,这是一种从 ChIP-seq 数据中确定转录因子结合位置的计算方法。CSDeconv 与先前的方法不同,它使用盲去卷积方法,可以准确地调用紧密间隔的结合位点。我们将 CSDeconv 应用于新型结核分枝杆菌 DosR 结合的 ChIP-seq 数据和现有的人类 GABP 数据,并表明它可以区分相隔仅 40bp 的结合位点。