Graduate School of Information Sciences, Tohoku University, Sendai, Miyagi, Japan.
Advanced Research Laboratory, Canon Medical Systems Corporation, Otawara, Tochigi, Japan.
BMC Bioinformatics. 2020 Sep 22;21(1):417. doi: 10.1186/s12859-020-03729-6.
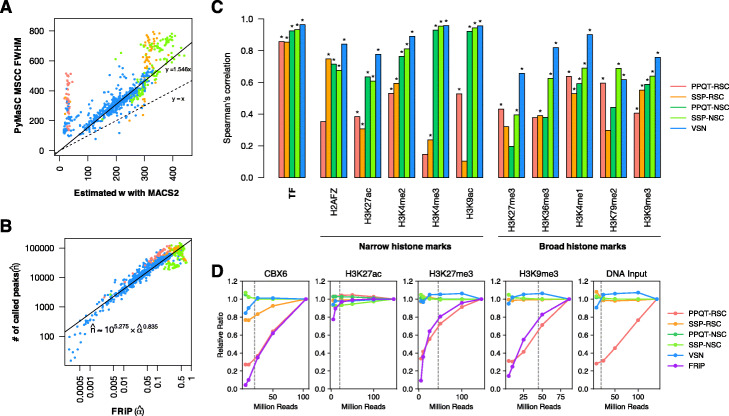
Strand cross-correlation profiles are used for both peak calling pre-analysis and quality control (QC) in chromatin immunoprecipitation followed by sequencing (ChIP-seq) analysis. Despite its potential for robust and accurate assessments of signal-to-noise ratio (S/N) because of its peak calling independence, it remains unclear what aspects of quality such strand cross-correlation profiles actually measure.
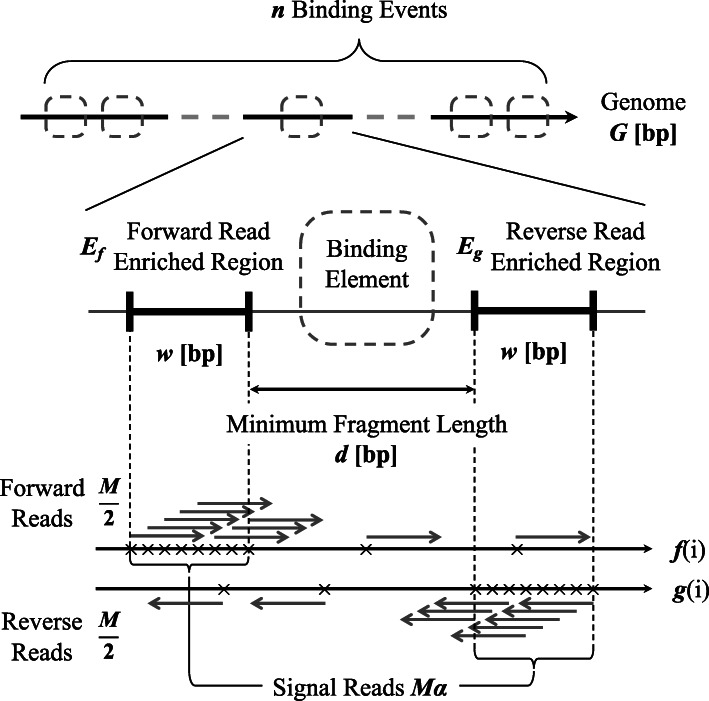
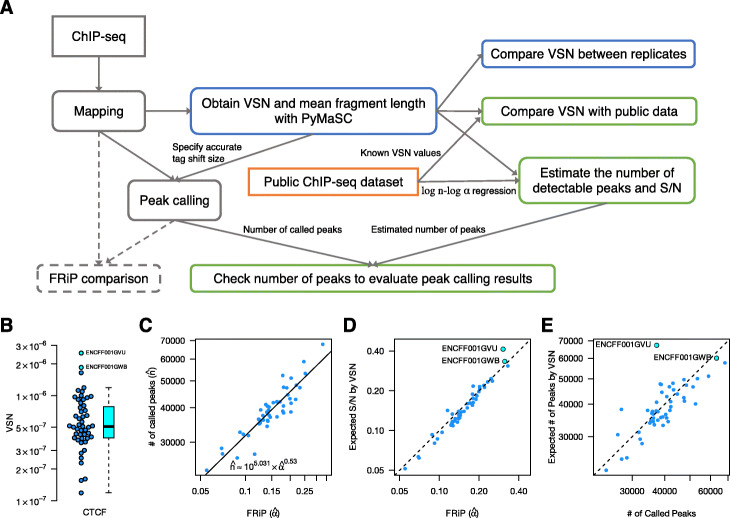
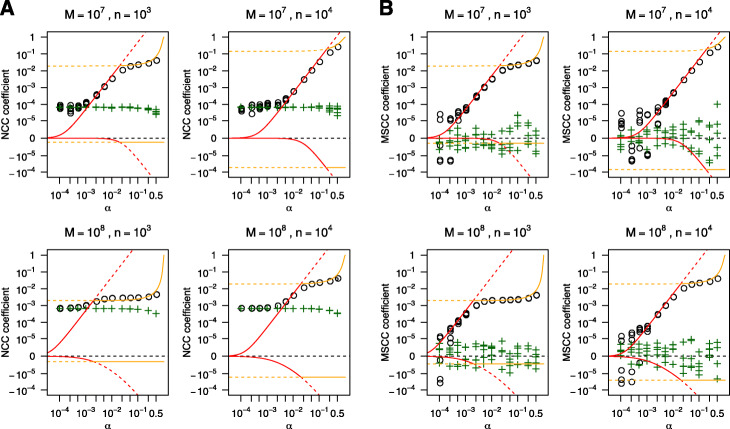
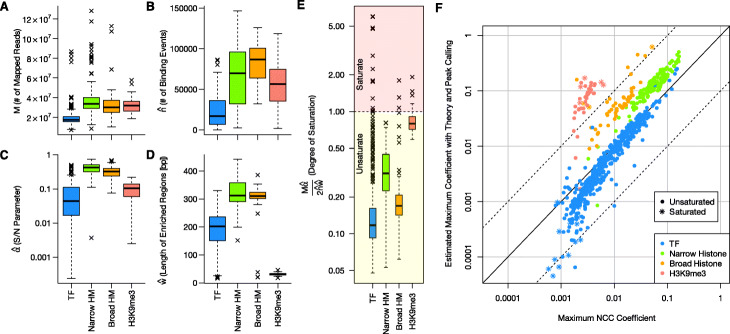
We introduced a simple model to simulate the mapped read-density of ChIP-seq and then derived the theoretical maximum and minimum of cross-correlation coefficients between strands. The results suggest that the maximum coefficient of typical ChIP-seq samples is directly proportional to the number of total mapped reads and the square of the ratio of signal reads, and inversely proportional to the number of peaks and the length of read-enriched regions. Simulation analysis supported our results and evaluation using 790 ChIP-seq data obtained from the public database demonstrated high consistency between calculated cross-correlation coefficients and estimated coefficients based on the theoretical relations and peak calling results. In addition, we found that the mappability-bias-correction improved sensitivity, enabling differentiation of maximum coefficients from the noise level. Based on these insights, we proposed virtual S/N (VSN), a novel peak call-free metric for S/N assessment. We also developed PyMaSC, a tool to calculate strand cross-correlation and VSN efficiently. VSN achieved most consistent S/N estimation for various ChIP targets and sequencing read depths. Furthermore, we demonstrated that a combination of VSN and pre-existing peak calling results enable the estimation of the numbers of detectable peaks for posterior experiments and assess peak calling results.
We present the first theoretical insights into the strand cross-correlation, and the results reveal the potential and the limitations of strand cross-correlation analysis. Our quality assessment framework using VSN provides peak call-independent QC and will help in the evaluation of peak call analysis in ChIP-seq experiments.
链间相关轮廓用于预分析峰调用和质量控制(QC),在染色质免疫沉淀测序(ChIP-seq)分析中。尽管由于其峰调用独立性,它具有稳健和准确评估信号噪声比(S/N)的潜力,但仍不清楚链间相关轮廓实际上测量了哪些质量方面。
我们引入了一个简单的模型来模拟 ChIP-seq 的映射读密度,然后推导出链间交叉相关系数的理论最大值和最小值。结果表明,典型 ChIP-seq 样本的最大相关系数与总映射读数量成正比,与信号读的比例的平方成正比,与峰的数量和读富集区域的长度成反比。模拟分析支持了我们的结果,并且使用来自公共数据库的 790 个 ChIP-seq 数据的评估表明,计算出的交叉相关系数与基于理论关系和峰调用结果的估计系数之间具有高度一致性。此外,我们发现可映射性偏差校正提高了灵敏度,使最大系数能够从噪声水平中区分出来。基于这些见解,我们提出了虚拟 S/N(VSN),一种用于 S/N 评估的新型无峰调用指标。我们还开发了 PyMaSC,一种有效计算链间交叉相关和 VSN 的工具。VSN 实现了对各种 ChIP 靶标和测序读深度的最一致的 S/N 估计。此外,我们证明了 VSN 和现有的峰调用结果的组合能够估计后续实验中可检测峰的数量,并评估峰调用结果。
我们首次提出了链间相关的理论见解,结果揭示了链间相关分析的潜力和局限性。我们使用 VSN 的质量评估框架提供了无峰调用的 QC,并将有助于评估 ChIP-seq 实验中的峰调用分析。